Is it an orbital foreign body?
Hong
Kong Med J 2017 Dec;23(6):653.e1–2
DOI: 10.12809/hkmj164952
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Is it an orbital foreign body?
Mohamed Shaheeda, FRCSEd, MPH; Stacey C Lam, MB,
ChB; Noel CY Chan, FRCSEd, FCOphthHK; Hunter KL Yuen, FRCSEd, FCOphthHK
Department of Ophthalmology and Visual Sciences,
The Chinese University of Hong Kong, Shatin, Hong Kong
Corresponding author: Dr Stacey C Lam (staceylam@gmail.com)
 Full paper in PDF
Full paper in PDF
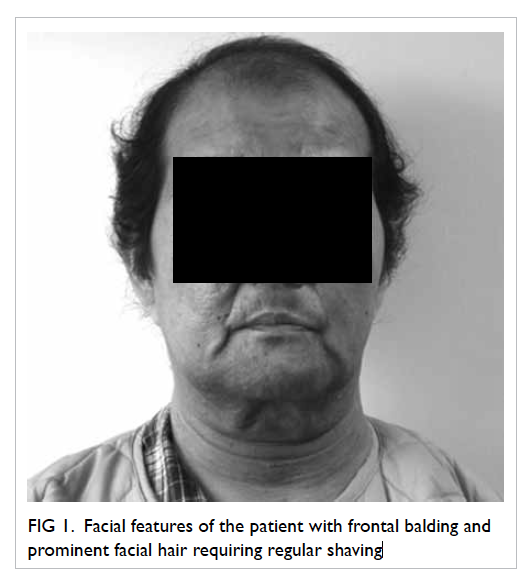
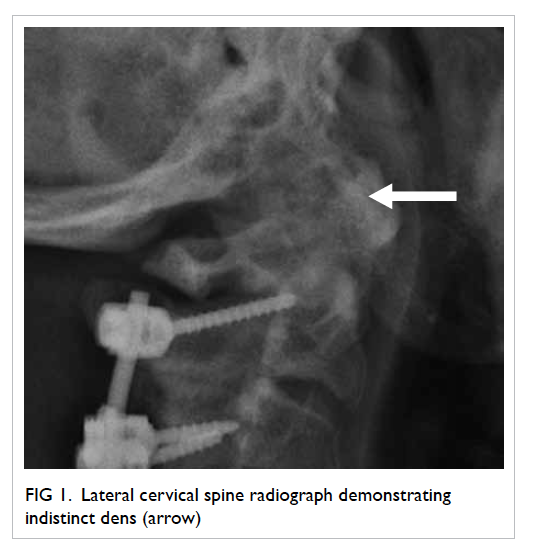
A 58-year-old man presented with left globe rupture
following blunt orbital trauma. Visual acuity was light perception only.
There was lid swelling, ecchymosis, and a deformed globe with scleral
laceration. Computed tomography (CT) of the orbit revealed a 3.7-mm
hyperdensity at the superomedial aspect of the left globe, interpreted by
the radiologist as a possible intra-orbital foreign body (Fig).
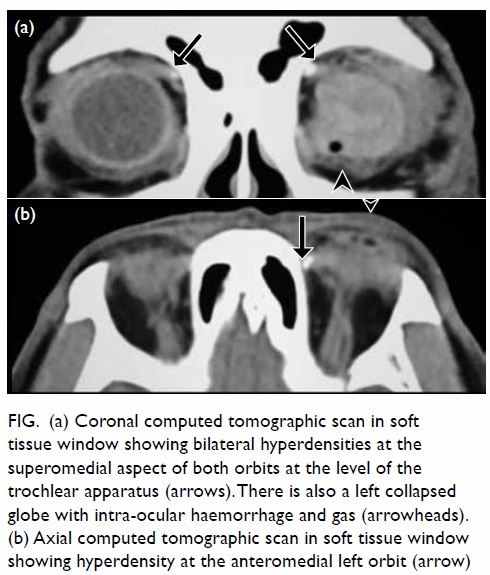
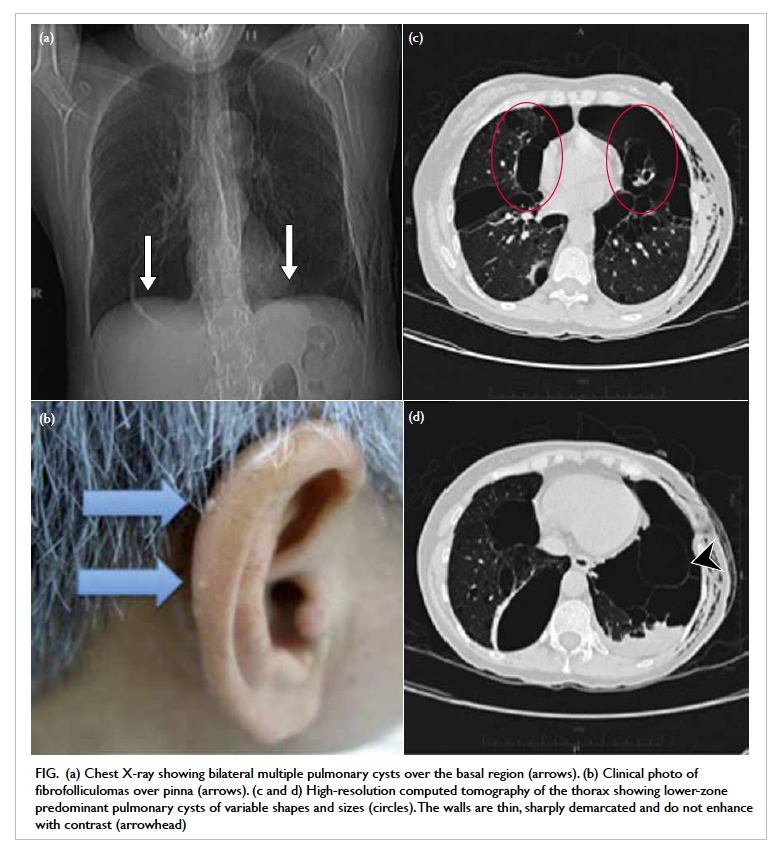
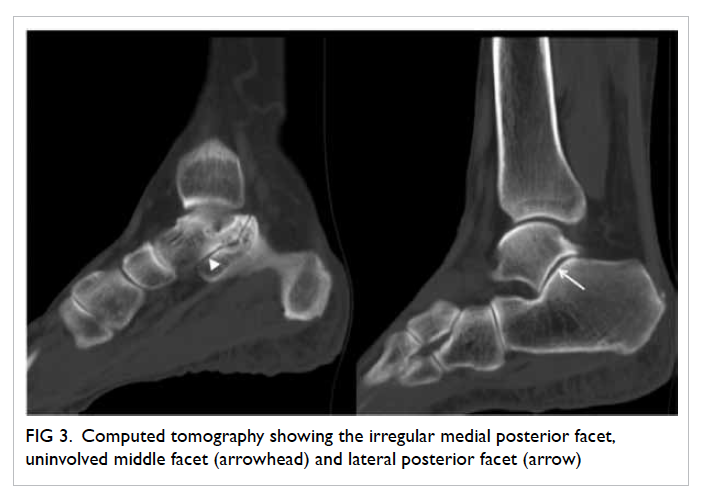
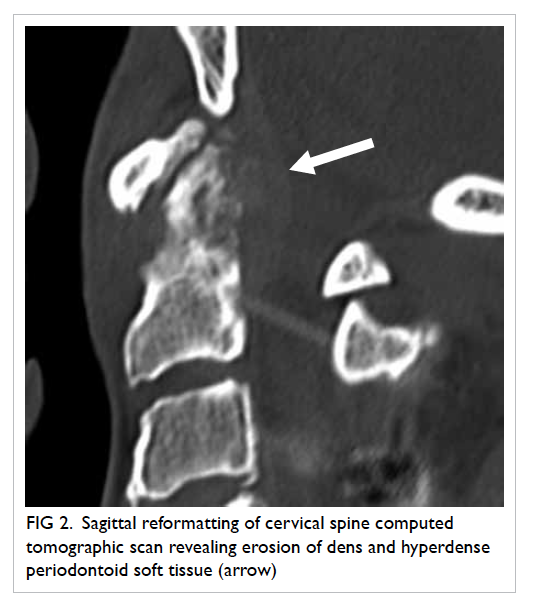
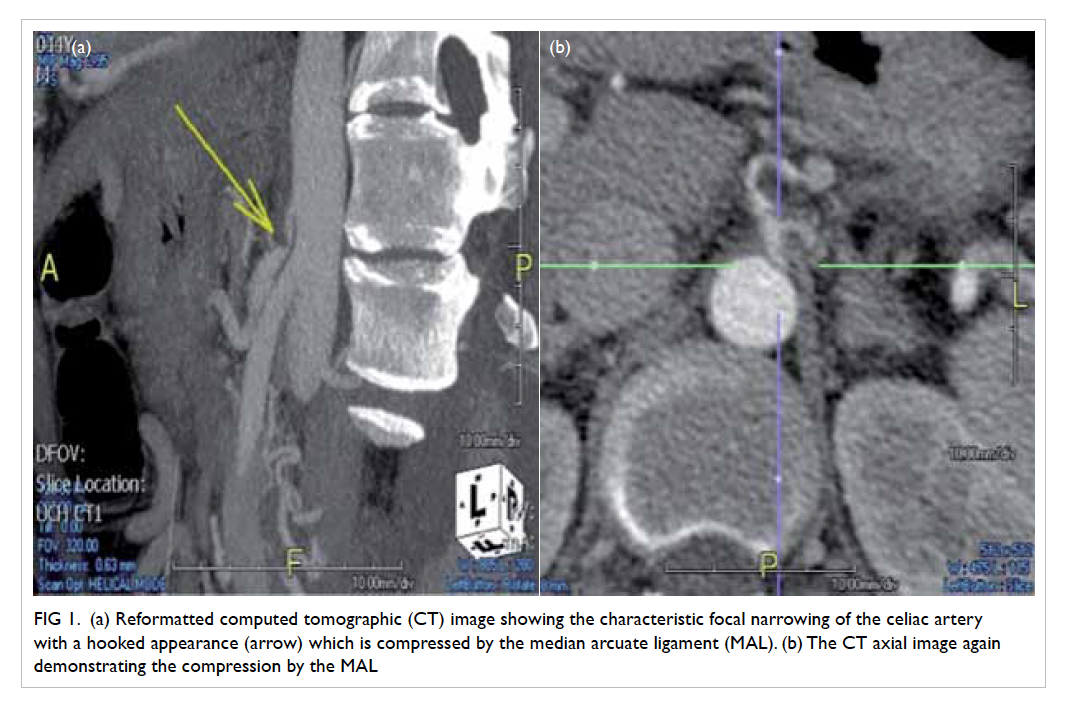
Figure. (a) Coronal computed tomographic scan in soft tissue window showing bilateral hyperdensities at the superomedial aspect of both orbits at the level of the trochlear apparatus (arrows). There is also a left collapsed globe with intra-ocular haemorrhage and gas (arrowheads). (b) Axial computed tomographic scan in soft tissue window showing hyperdensity at the anteromedial left orbit (arrow)
Thorough history for the mechanism of trauma,
physical examination for a wound site and thin cut CT sections were
reviewed. History revealed blunt orbital trauma by a metallic rod rather
than a sharp penetrating injury. No cutaneous entry site was evident.
Computed tomography revealed bilateral hyperdensities at the level of the
trochlea, more pronounced on the left than the right, with no metallic
streak artefacts. All of the above supported a diagnosis of trochlear
calcification rather than an intra-orbital foreign body, and the patient
subsequently underwent emergency repair of scleral laceration without
orbital exploration.
Discussion
The trochlea is a cartilaginous pulley at the
superomedial aspect of the orbit through which the superior oblique muscle
tendon passes freely. It has a synovium-lined space, and like other
synovial joints of the body, the trochlea can develop calcifications in
the cartilage, tendon, or within the bursa-like cleft.1 The reported prevalence of incidental trochlear
calcifications on CT is 3-16%, with over 50% being unilateral.2 3 No clear
cause has been identified, but prior studies have postulated degenerative,
inflammatory, metabolic or traumatic aetiologies.4
Since trochlear calcification is asymptomatic, most cases go unnoticed by
radiologists as well as ophthalmologists. In the presence of co-existing
orbital trauma, it can be misdiagnosed as an intraorbital foreign body.
Surgical exploration around the trochlear region may cause damage and
scarring leading to diplopia.
Differentiation of the two entities requires
history taking, physical examination, and proper imaging. A foreign body
is present in one in six cases of orbital trauma, with metallic objects
and glass being the most common.5 A
review of the history to determine mechanism of injury is useful but may
be unreliable. Examination of the skin and conjunctiva, particularly the
fornices, may help detect subtle penetrating injuries. Radiological
assessment includes plain films, ultrasound, CT, and magnetic resonance
imaging. Plain films are limited to detection of metallic foreign bodies,
and ultrasound has limited use because foreign bodies can be masked by
surrounding highly reflective structures such as bone. Computed tomography
is an excellent means to identify high-density objects such as metal or
glass, but not organic matter. Magnetic resonance imaging is
contra-indicated in metallic foreign bodies, but may be useful for organic
foreign matter. Features of trochlear calcification may include
symmetrical presentation and typical site at the trochlear apparatus. Its
morphology has been described as comma, dot, and inverted U shape.2 In contrast, orbital foreign bodies are often
unilateral with no specific size, shape, or location. Metallic foreign
bodies may also generate streak artefacts.
Orbital calcifications can be incidental or
pathological. It is important to recognise trochlear calcifications as
distinct from foreign bodies so as to avoid unnecessary surgical
exploration in the presence of orbital trauma.
References
1. Sobel RK, Goldstein SM. Trochlear
calcification: A common entity. Orbit 2012;31:94-6. Crossref
2. Xiao TL, Kalariya NM, Yan ZH, et al.
Trochlear calcification and intraorbital foreign body in ocular trauma
patients. Chin J Traumatol 2009;12:210-3.
3. Shriver EM, McKeown CA, Johnson TE.
Trochlear calcification mimicking an orbital foreign body. Ophthal Plast
Reconstr Surg 2011;27:143-4. Crossref
4. Buch K, Nadgir RN, Tannenbaum AD,
Ozonoff A, Fujita A, Sakai O. Clinical significance of trochlear
calcifications in the orbit. AJNR Am J Neuroradiol 2014;35:573-7. Crossref
5. Nasr AM, Haik BG, Fleming JC, Al-Hussain
HM, Karcioglu ZA. Penetrating orbital injury with organic foreign bodies.
Ophthalmology 1999;106:523-32. Crossref