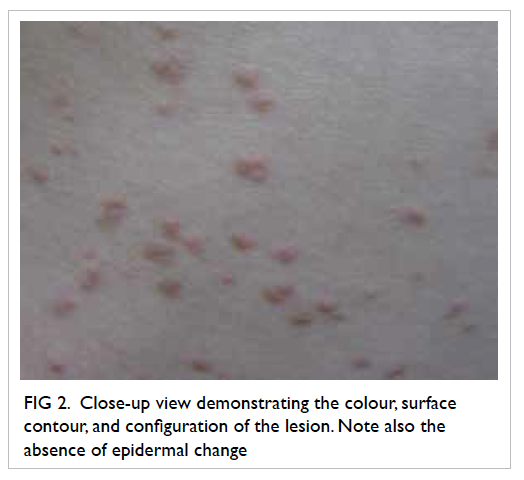
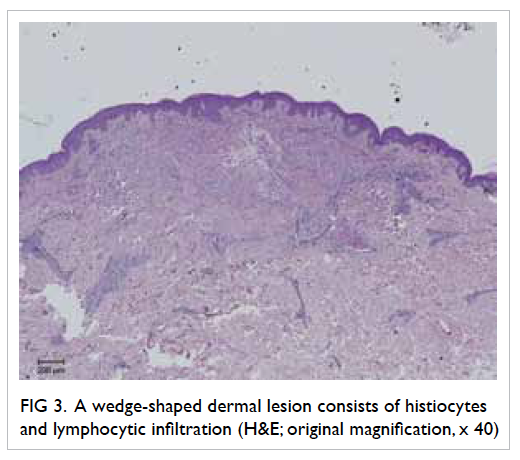
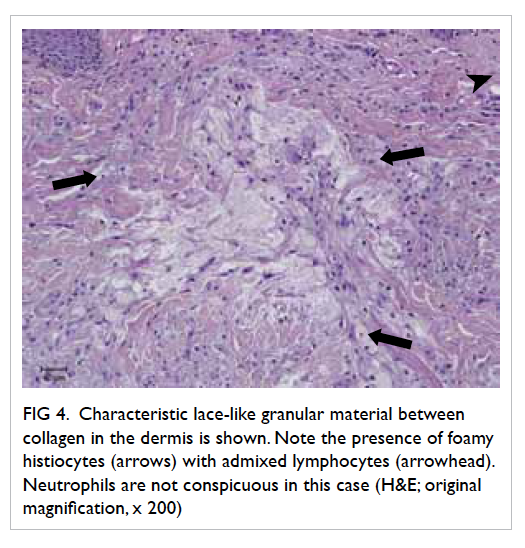
An uncommon complication of infective endocarditis
Hong Kong Med J 2015 Apr;21(2):187.e1–2
DOI: 10.12809/hkmj144248
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
An uncommon complication of infective endocarditis
KW Lam, MB, BS, FHKAM (Medicine); KW Au Yeung, MB, BS, FHKAM (Anaesthesiology); KY Lai, MB, BS, FHKAM (Medicine)
Intensive Care Unit, Queen Elizabeth Hospital, Jordan, Hong Kong
Corresponding author: Dr KW Lam (lamkw1@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
A 20-year-old man presented to us with confusion and
generalised skin rash in April 2010. On examination,
he was febrile and in shock. He was detected with a
mitral regurgitation murmur and mild neck stiffness.
His white cell count was elevated and platelet count
was low. His liver function was mildly impaired.
Chest X-ray showed acute pulmonary oedema.
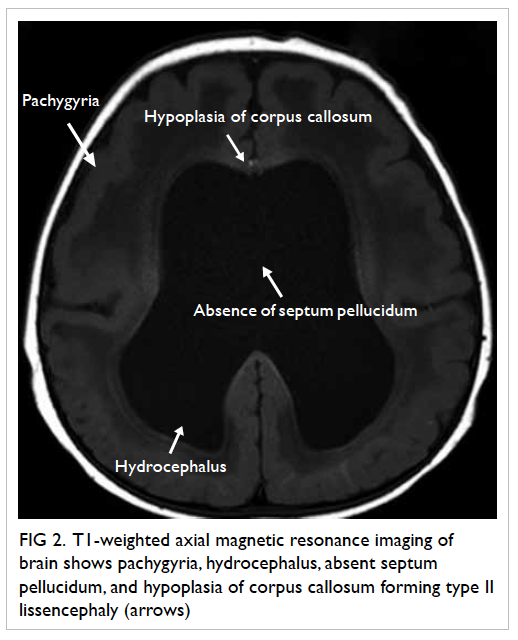
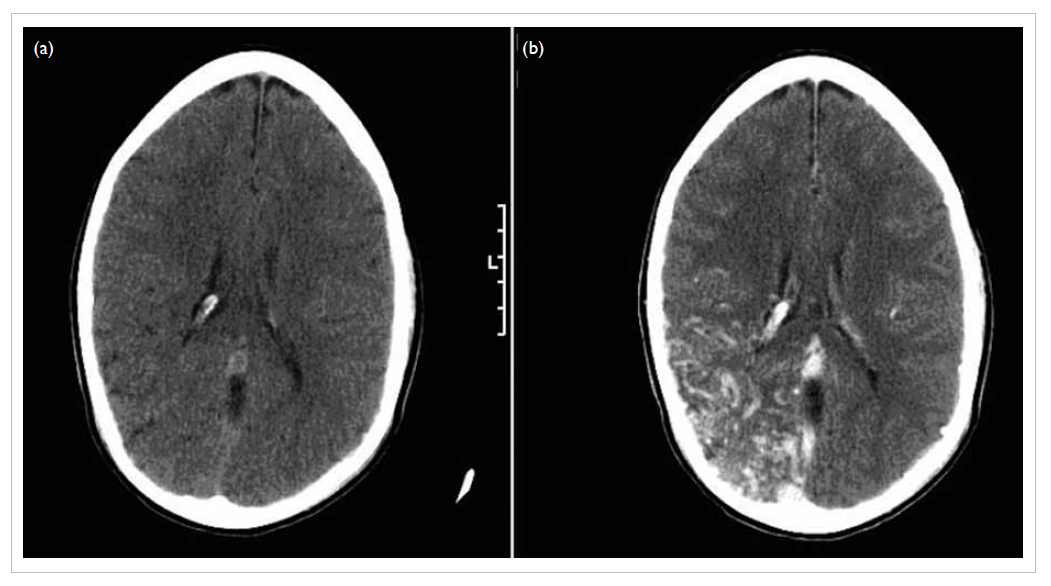
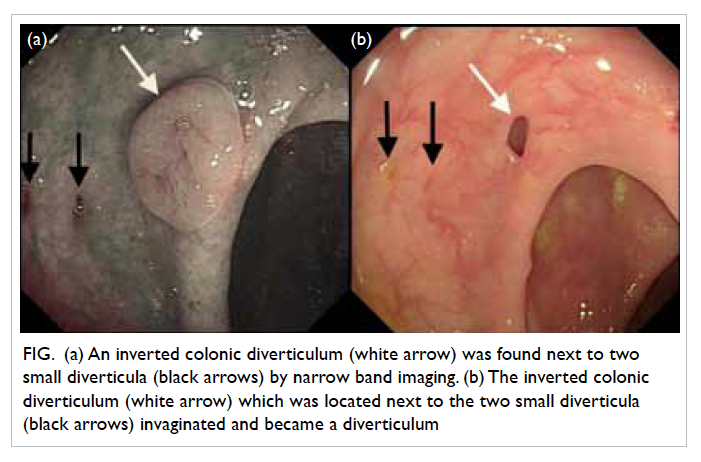
Computed tomography (CT) scan of brain showed
multiple haematomas in his left occipital lobe and
right parietal lobe, and subarachnoid haemorrhage
(Fig 1a). Blood culture showed methicillin-sensitive
Staphylococcus aureus (MSSA).
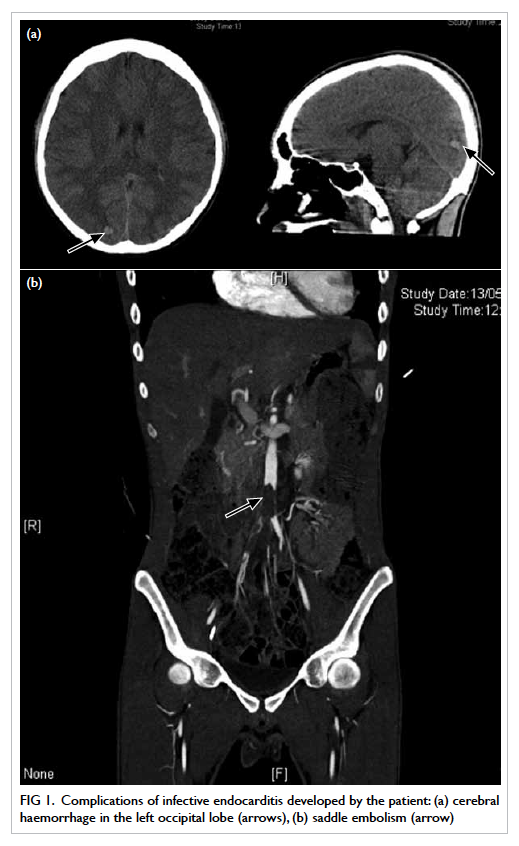
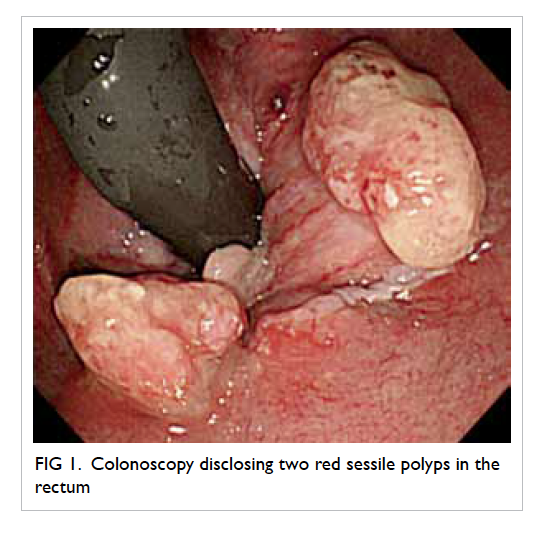
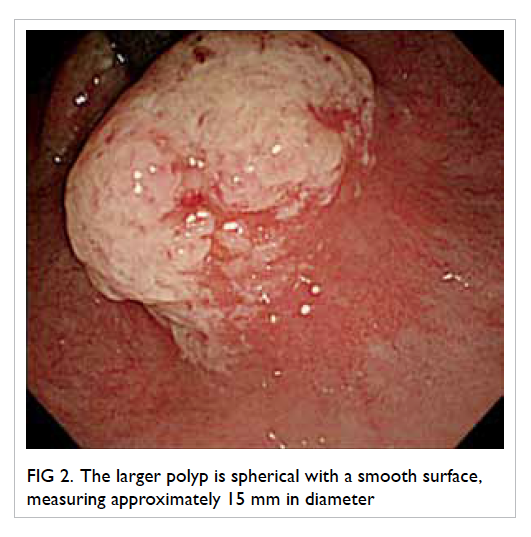
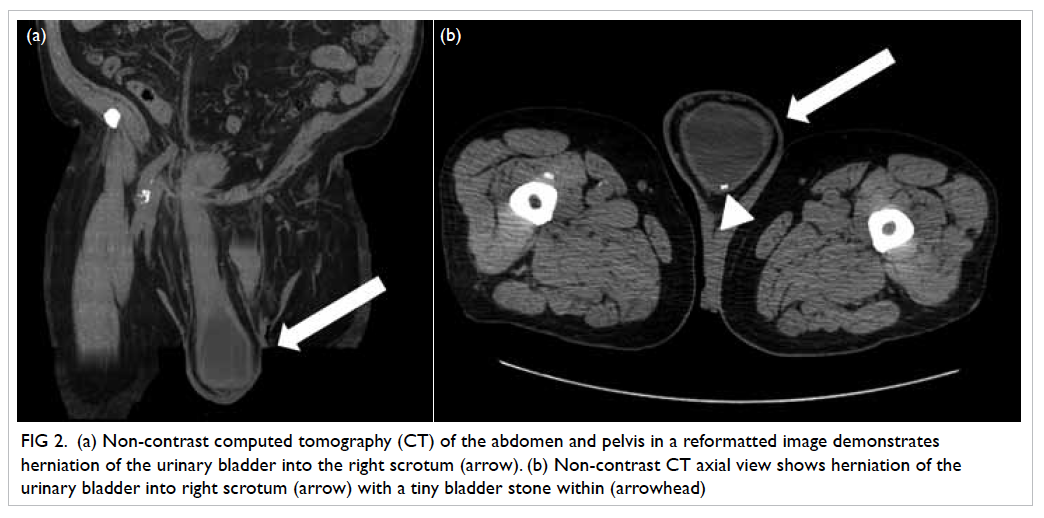
Figure 1. Complications of infective endocarditis developed by the patient: (a) cerebral haemorrhage in the left occipital lobe (arrows), (b) saddle embolism (arrow)
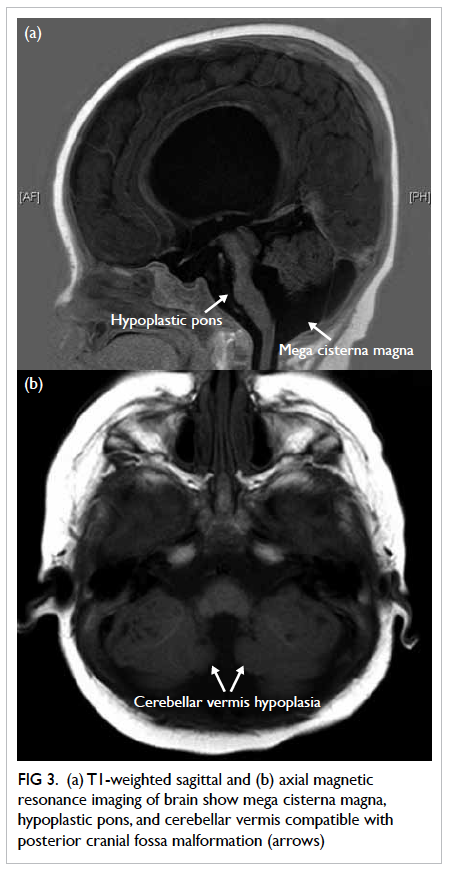
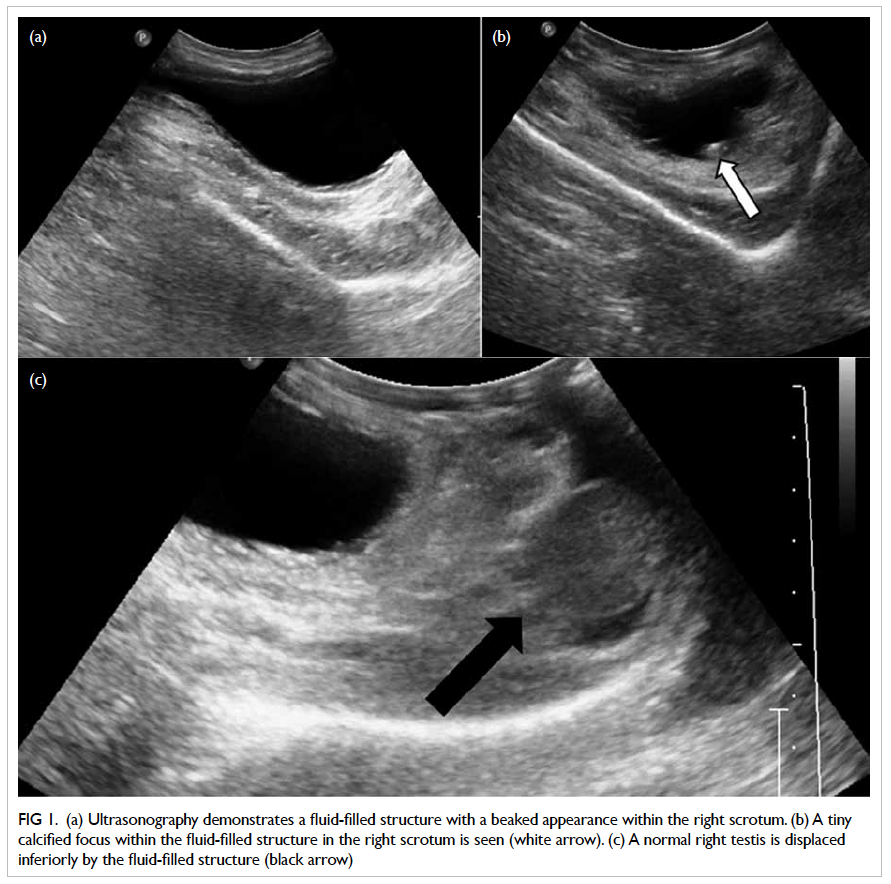
Transthoracic echocardiogram showed a huge
vegetation measuring 4 cm x 2.22 cm attached to
the base of anterior leaflet of mitral valve, resulting
in perforation of the base of leaflet with severe
mitral regurgitation (Fig 2a). The diagnosis was
infective endocarditis due to MSSA, complicated by
ruptured chordae tendineae with severe acute mitral
regurgitation and multiple, septic cerebral emboli.
The source of infection was suspected to be the skin.
He was treated with high-dose intravenous cloxacillin
and gentamicin. His heart failure was treated with
frusemide.
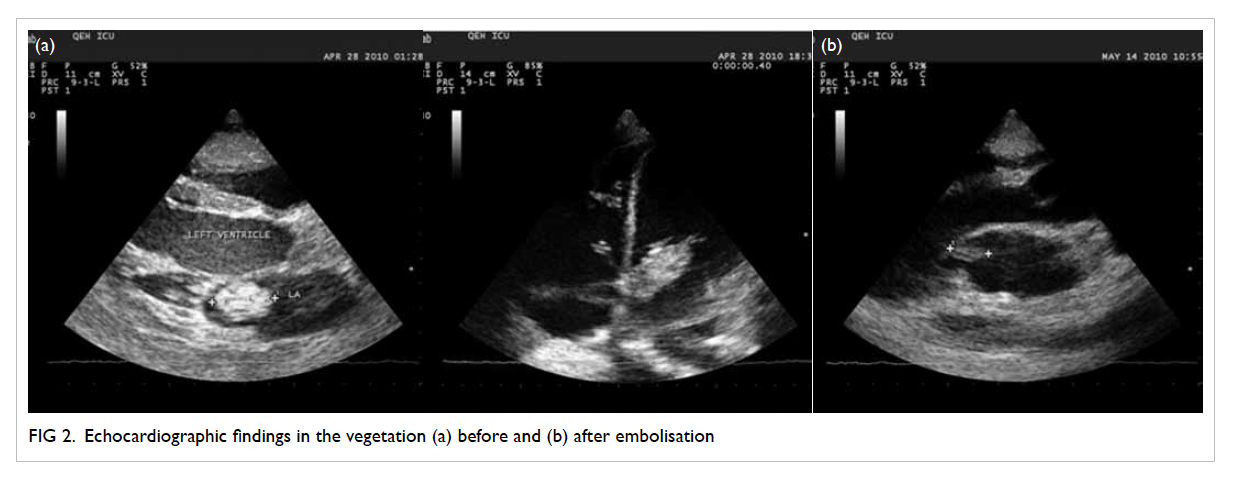
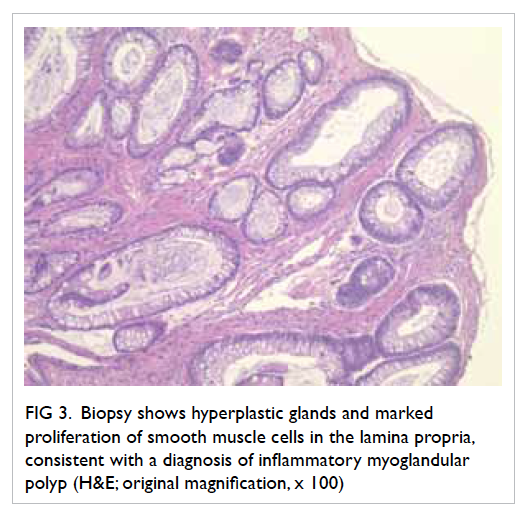
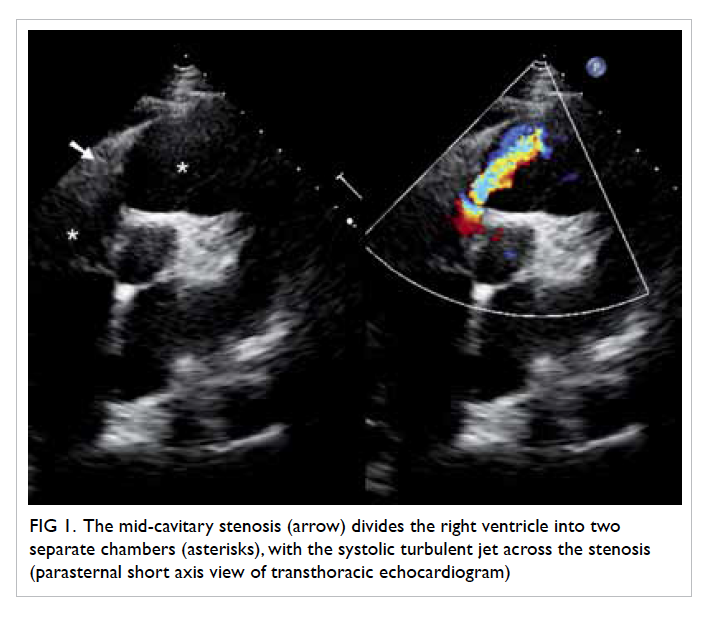
Figure 2. Echocardiographic findings in the vegetation (a) before and (b) after embolisation
Three weeks later, he complained of sudden
onset of right lower limb pain and numbness. His
CT angiogram showed a large saddle embolus in the
lower abdominal aorta (Fig 1b). Emergent right-sided
lower femoral and bilateral iliac artery embolectomy
was performed.
On postoperative echocardiogram, the size of
the vegetation was found to be decreased to 1.5 cm in
diameter (Fig 2b). He then underwent mitral valvular
replacement about 1 week later.
Discussion
The incidence of community-acquired native-valve
endocarditis in western countries ranges from 1.7
to 6.2 cases per 100 000 person-years, with a male-to-female ratio of 1.7:1.1 Recently, the incidence of S
aureus infective endocarditis is on the rise. Infective
endocarditis due to S aureus is more common among
young adults, especially the intravenous injection
drug users.1 Usually, the tricuspid valve is involved.2
Extra cardiac complications of infective
endocarditis usually include embolic events. The
rate of embolic events has a relationship with the
initiation of antibiotic therapy. In one study, after
the commencement of appropriate antimicrobial
treatment, the rate of embolism fell from 13 per 1000
patient-days during the first week of treatment to
fewer than 1.2 per 1000 patient-days 2 weeks after
treatment.3 A review involving 281 patients with
suspected infective endocarditis demonstrated that
the incidence of embolic events was greater with
mitral than aortic valve vegetations (25% vs 10%).4
For mitral valve vegetations, the rate of embolism was
higher if these were attached to the anterior leaflet
rather than posterior leaflet. Some studies showed
that the rate of embolism correlated with the size of
vegetation, with the risk being higher if the diameter
of the vegetation was greater than 1 cm.1
Up to 65% of embolic events of infective
endocarditis are associated with neurological
involvement. Such neurological complications
account for 20% to 40% of all patients with infective
endocarditis.5 Saddle embolus is a large embolus that
straddles the arterial bifurcation and, thus, blocks
both branches of the aorta. Saddle embolisation at
the aortic bifurcation is an uncommon but serious
complication. From the literature search, only eight
cases were reported and all were caused by fungal
endocarditis.6
References
1. Mylonakis E, Calderwood SB. Infective endocarditis in
adults. N Engl J Med 2001;345:1318-30. Crossref
2. Hecht SR, Berger M. Right-sided endocarditis in
intravenous drug users. Prognostic features in 102
episodes. Ann Intern Med 1992;117:560-6. Crossref
3. Heiro M, Nikoskelainen J, Engblom E, Kotilainen E,
Marttila R, Kotilainen P. Neurologic manifestations of
infective endocarditis: a 17-year experience in a teaching
hospital in Finland. Arch Inern Med 2000;160:2781-7. Crossref
4. Rohmann S, Erbel R, Görge G, et al. Clinical relevance of vegetation
localization by transoesophageal echocardiography in
infective endocarditis. Eur Heart J 1992;13:446-52.
5. Røder BL, Wandall DA, Espersen F, Frimodt-Møller
N, Skinhøj P, Rosdahl VT. Neurologic manifestations
in Staphylococcus aureus endocarditis: a review of
260 bacteremic cases in nondrug addicts. Am J Med
1997;102:379-86. Crossref
6. Kawamoto T, Nakano S, Matsuda H, Hirose H, Kawashima
Y. Candida endocarditis with saddle embolism: a
successful surgical intervention. Ann Thorac Surg
1989;48:723-4. Crossref