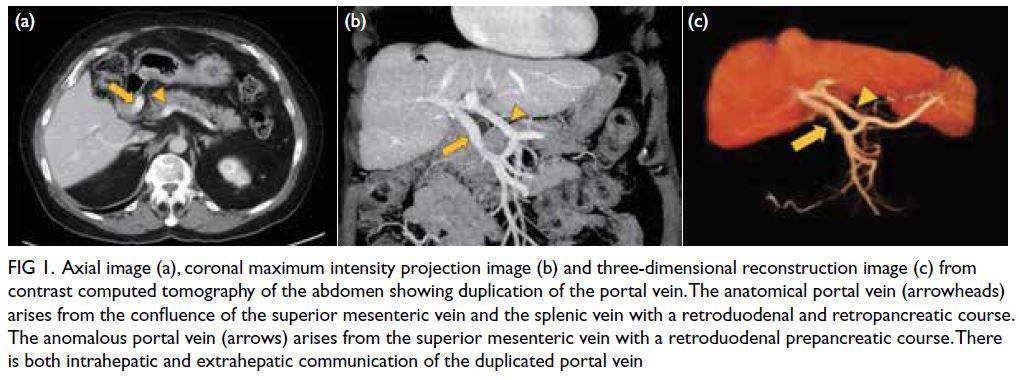
Dermatomyositis following COVID-19 vaccination
Hong Kong Med J 2025 Feb;31(1):74.e1–2 | Epub 10 Feb 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Dermatomyositis following COVID-19 vaccination
TK Kong, FRCP, FHKAM (Medicine)
Department of Medicine and Therapeutics, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong SAR, China
Corresponding author: Dr TK Kong (tkkong@cuhk.edu.hk)
 Full paper in PDF
Full paper in PDF
A 59-year-old Chinese woman, a workman with
hypertension and non-smoking history, sought
medical advice in April 2024 for a 16-month
history of progressive weight loss, reduced appetite,
proximal myalgia, arthralgia of the hands, abdominal
discomfort and constipation since February 2023;
and a 10-month history of dry cough and exertional
dyspnoea. She had received four Comirnaty
messenger ribonucleic acid coronavirus disease 2019
(COVID-19) vaccinations (Pfizer-BioNTech; Pfizer
Inc, Philadelphia [PA], United States) between 8 July
2021 and 12 January 2023 and had two COVID-19
infections, one each in March 2022 and June 2023.
She carried the beta thalassaemia trait, as did her
father. Both her parents had lung cancer.
Investigations in January 2024 revealed raised
carcinoembryonic antigen level (11.4 ug/L; reference
range, <5.0); negative stool for occult blood; negative
sputum for acid-fast bacilli smear/culture and
positive anti–nuclear antigen antibodies (1:160).
Whole-body positron emission tomography–computed tomography in January 2024 revealed
patchy ground-glass opacities and fibrosis with mild 18F-fluorodeoxyglucose uptake in the lower lobes of
the lungs, but no hypermetabolic lesion to suggest
malignancy. Twelve days prior to consultation she
had been hospitalised for dizziness and found to
have a low haemoglobin level (6.7 g/dL). She was
transfused to 8.5 g/dL and discharged the next day.
When seen at the clinic in April 2024, the
patient’s body weight was 47.9 kg, compared
with 68.2 kg 16 months previously. She expressed
difficulty with working and in getting up from
bed because of weakness. Physical examination
revealed pallor, a rash on her hands suggestive
of dermatomyositis (Fig), puffy eyelids without
heliotrope rash, and proximal muscle wasting and
weakness. Fine end-inspiratory crepitations were
heard at the base of the lungs. There was no cervical
lymphadenopathy nor palpable abdominal masses.
Review showed a serial drop in haemoglobin level
(from 10.2 g/dL in February 2023 to 6.7 g/dL in
April 2024), increasing microcytosis (decrease in
mean corpuscular volume from 63.1 fL in February
2023 to 59.0 fL in April 2024), and iron deficiency
in the setting of chronic inflammation. The clinical
diagnosis was dermatomyositis with myopathy,
interstitial lung disease, and probable colon cancer
with iron deficiency anaemia superimposed on
beta thalassaemia trait. She was referred to hospital
for further management. Muscle enzyme tests
revealed normal creatine kinase level (113 U/L)
but elevated lactate dehydrogenase (499 U/L) and
alanine aminotransferase levels (65 U/L). Myositis
antibody screening confirmed the diagnosis of
anti–melanoma differentiation–associated protein 5
(anti-MDA5) antibody–positive dermatomyositis.
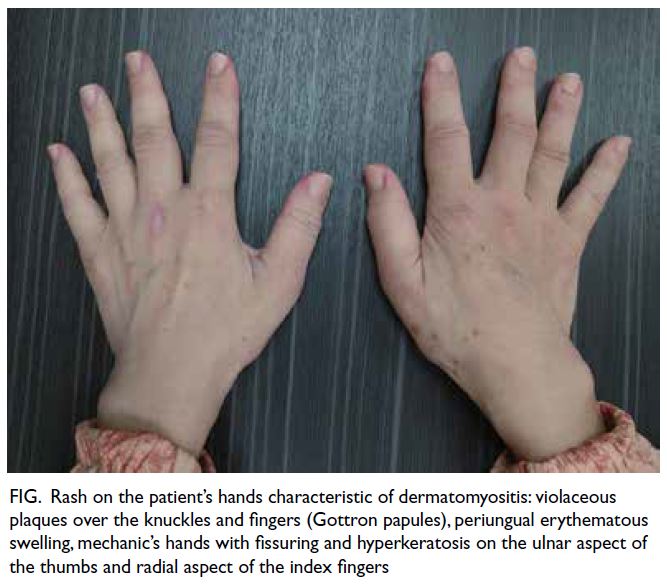
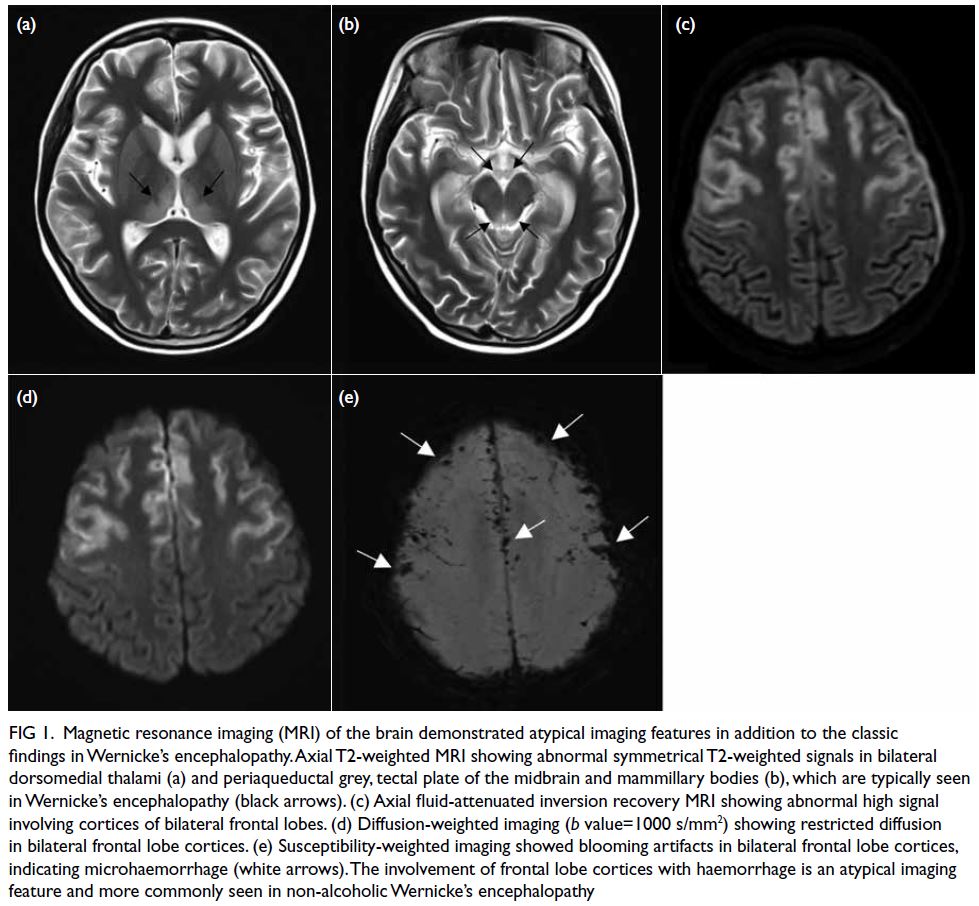
Figure. Rash on the patient’s hands characteristic of dermatomyositis: violaceous plaques over the knuckles and fingers (Gottron papules), periungual erythematous swelling, mechanic’s hands with fissuring and hyperkeratosis on the ulnar aspect of the thumbs and radial aspect of the index fingers
The patient’s illness onset in February 2023
following COVID-19 vaccination the month
before suggested a trigger by vaccination, further
aggravated by her COVID-19 infection in June
2023. Dermatomyositis, inflammatory myopathy,
and rheumatic immune-mediated inflammatory
diseases have been reported following COVID-19
vaccination and infection.1 2 3 In a systematic review
up to May 2023,3 24 cases of post–COVID-19
vaccination dermatomyositis were reported
worldwide, the majority following vaccination with
Pfizer-BioNTech vaccine, and some following that
with Moderna and Oxford–AstraZeneca vaccines.
Only two such cases were reported among Chinese,
one from mainland China (after Sinopharm’s
inactivated Vero cell),4 one from Taiwan (after Oxford–AstraZeneca),5 but none from Hong Kong.
The close temporal sequence and surge against
a background of the reported case series suggest
an association between severe acute respiratory
syndrome coronavirus 2 infection/vaccination and
the development of dermatomyositis, although
proof of causality requires further research because
of the limited number of cases reported.1 3 A recent
bioinformatic study and transcriptome-derived
insights point to a potential causal link between the
surge in the Yorkshire region in the United Kingdom
between 2020 and 2022 in anti-MDA5–positive
dermatomyositis, autoimmune interstitial lung
disease and COVID-19.6 The COVID-19 vaccination
and infection may trigger a proinflammatory
immune response involving type I interferon and
stimulate production of dermatomyositis-specific
autoantibodies such as MDA5 that are closely related
to viral defence or viral RNA interaction supporting
the concept of infection and vaccination-associated
dermatomyositis.1 2 3 6
This case demonstrates that dermatomyositis
can be induced by COVID-19 vaccination, ignorance
of which and of the diagnostic clinical signs
would lead to delayed diagnosis and management.
Coronavirus disease 2019 vaccines are widely used.
When patients present with constitutional symptoms
with persistent muscle aches and weakness following
COVID-19 vaccination, clinicians should consider
dermatomyositis as a differential diagnosis and
examine the skin for pathognomonic signs.
Author contributions
The author is solely responsible for the concept or design,
acquisition of data, analysis or interpretation of data, drafting
of the manuscript, and critical revision of the manuscript for
important intellectual content. The author had full access to
the data, contributed to the study, approved the final version
for publication, and takes responsibility for its accuracy and
integrity.
Conflicts of interest
The author has no conflicts of interest to disclose.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration
of Helsinki. Patient consent was obtained for clinical photo of
her hands and for publication of the article.
References
1. Holzer MT, Krusche M, Ruffer N, et al. New-onset
dermatomyositis following SARS-CoV-2 infection
and vaccination: a case-based review. Rheumatol Int
2022;42:2267-76. Crossref
2. Ding Y, Ge Y. Inflammatory myopathy following
coronavirus disease 2019 vaccination: a systematic review.
Front Public Health 2022;10:1007637. Crossref
3. Nune A, Durkowski V, Pillay SS, et al. New-onset rheumatic
immune-mediated inflammatory diseases following SARS-CoV-2 vaccinations until May 2023: a systematic review.
Vaccines (Basel) 2023;11:1571. Crossref
4. Yang L, Ye T, Liu H, Huang C, Tian W, Cai Y. A case of
anti-MDA5–positive dermatomyositis after inactivated
COVID-19 vaccine. J Eur Acad Dermatol Venereol
2023;37:e127-9. Crossref
5. Huang ST, Lee TJ, Chen KH, et al. Fatal myositis,
rhabdomyolysis and compartment syndrome after
ChAdOx1 nCoV-19 vaccination. J Microbiol Immunol
Infect 2022;55:1131-3. Crossref
6. David P, Sinha S, Iqbal K, et al. MDA5-autoimmunity
and interstitial pneumonitis contemporaneous with the
COVID-19 pandemic (MIP-C). EBioMedicine
2024:104:105136. Crossref