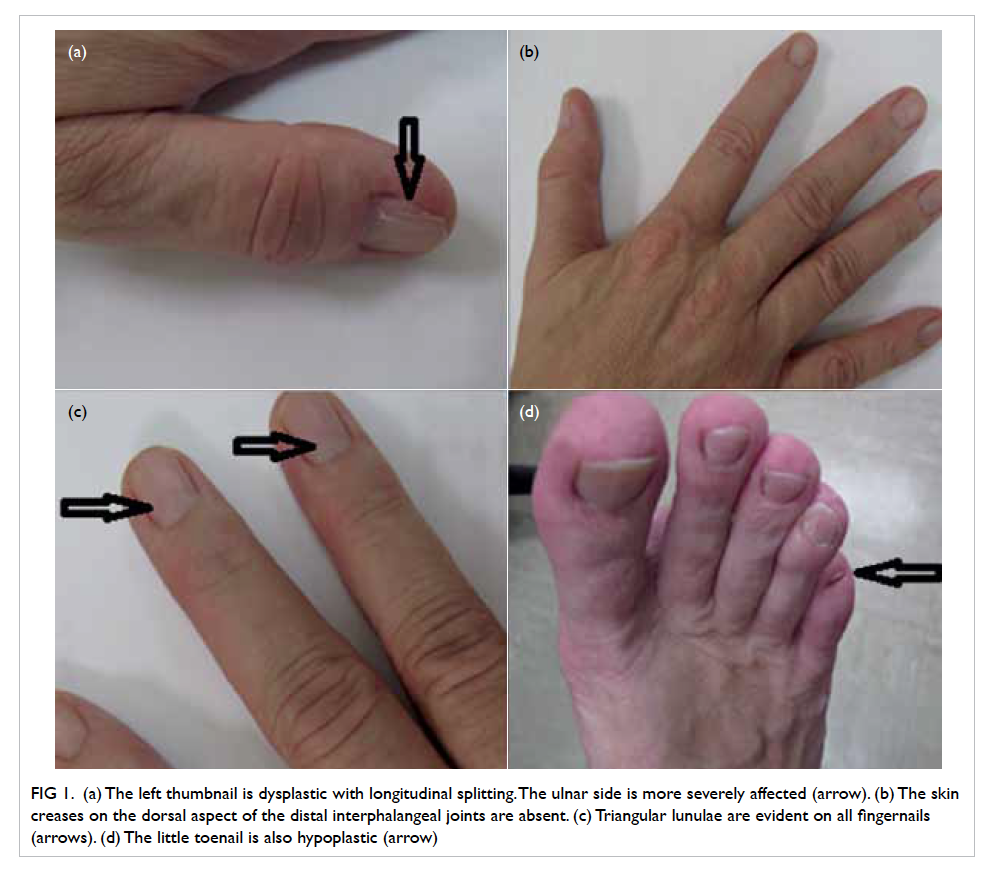
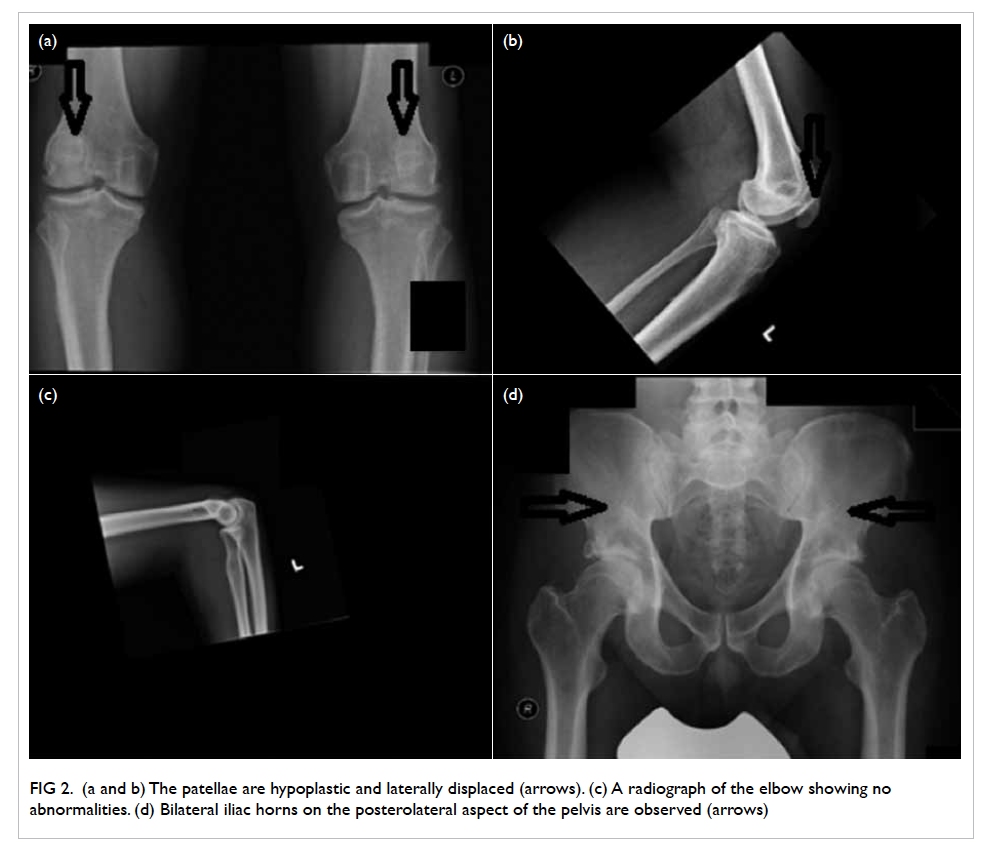
Lemierre’s syndrome: an often forgotten but potentially life-threatening disease
Hong Kong Med J 2016 Apr;22(2):184.e1–2
DOI: 10.12809/hkmj154696
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Lemierre’s syndrome: an often forgotten but potentially life-threatening disease
C Lee, MB, BS, FRCR;
Lorraine HY Sinn, MB, BS, FRCR;
Sonia HY Lam, MB, BS, FRCR;
WM Lam, MB, BS, FRCR
Department of Radiology, Queen Mary Hospital, Pokfulam, Hong Kong
Corresponding author: Dr C Lee (leechunbruce@gmail.com)
An earlier version of this paper was presented as a poster at the European
Society of Thoracic Radiology held in Barcelona, Spain on 4-6 June 2015.
 Full
paper in PDF
Full
paper in PDF
A 20-year-old Chinese female with good past health
presented to the emergency department in October
2014 with a few days history of fever, dizziness, and
headache. She was hypotensive upon presentation
with blood pressure of 73/48 mm Hg and pulse rate
of 90 beats/min but responded to fluid resuscitation.
Physical examination revealed no other significant
findings. Her white cell count was 4.4 x 109 /L on
admission but increased to 13.4 x 109 five days later.
Neutrophil predominance was observed. Subsequent
gradual decline and normalisation of the white cell
count was noted after initiation of intravenous
antibiotics.
Computed tomographic (CT) abdomen and
pelvis was initially requested based on the clinical
suspicion of intra-abdominal sepsis or gynaecological
pathologies but yielded no remarkable findings. Chest
X-ray (CXR) was also performed for preliminary
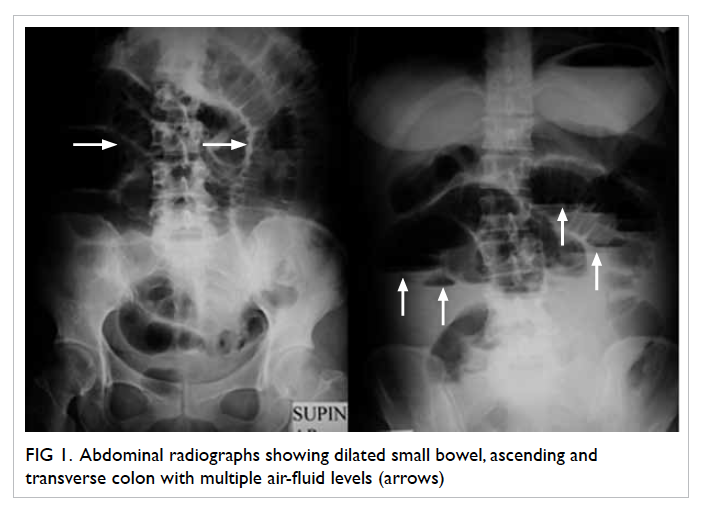
assessment and was initially unremarkable. Follow-up
serial CXRs, however, showed an increasing
number of cavitary lesions in both lungs with no
zonal predominance (Fig 1).
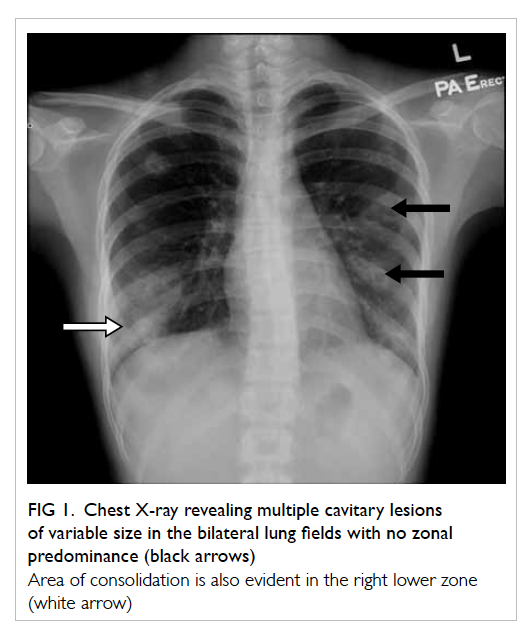
Figure 1. Chest X-ray revealing multiple cavitary lesions of variable size in the bilateral lung fields with no zonal predominance (black arrows)
Area of consolidation is also evident in the right lower zone (white arrow)
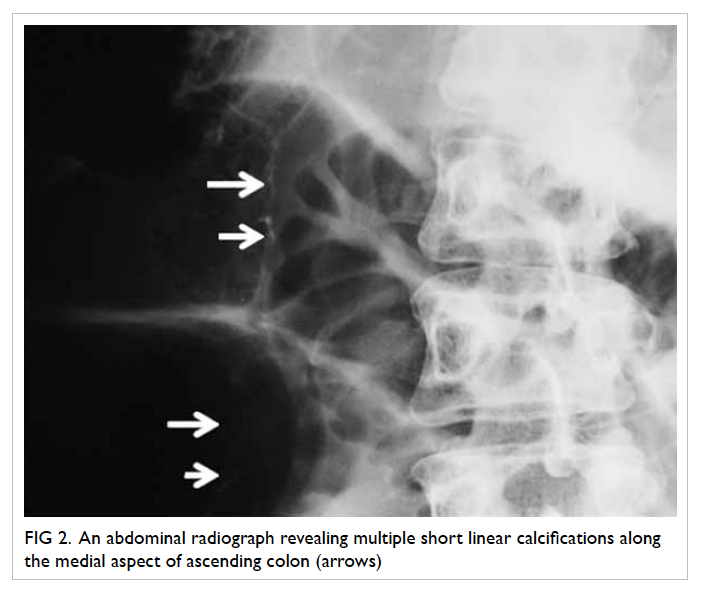
Non-contrast CT thorax was deemed necessary
for further characterisation of these lesions and
confirmed multiple cavitary lesions of variable sizes
involving all the lung lobes (Fig 2). Thick irregular
walls with fluid content and perifocal consolidative
changes were noted in some of these lesions, which are
compatible with an infective process. Further imaging
workup was then arranged to identify any potential
source of the septic emboli but echocardiogram did
not reveal any heart valve vegetation.
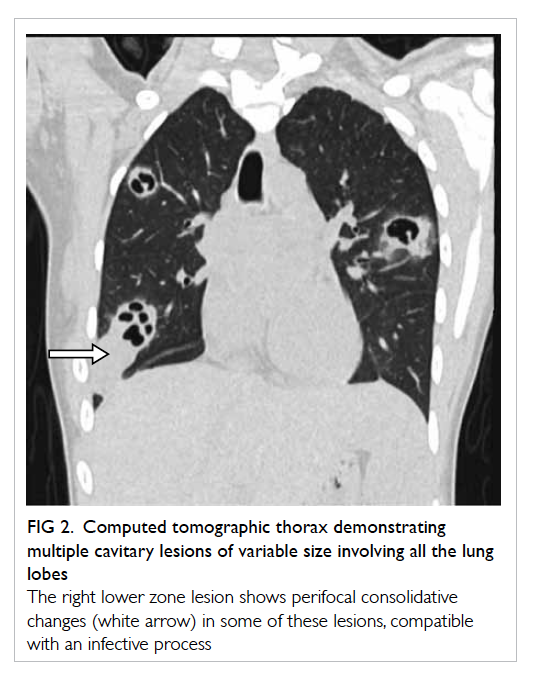
Figure 2. Computed tomographic thorax demonstrating multiple cavitary lesions of variable size involving all the lung lobes
The right lower zone lesion shows perifocal consolidative changes (white arrow) in some of these lesions, compatible with an infective process
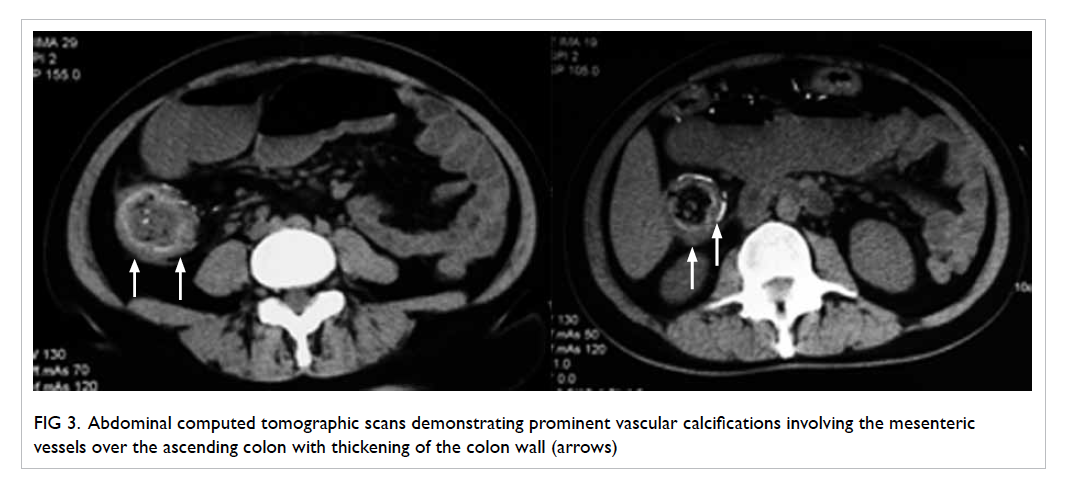
On further questioning, the patient reported
mild left neck pain. Doppler ultrasound of the neck
was then performed based on the clinical suspicion
of Lemierre’s syndrome. A markedly narrowed lumen
with thickened wall and dampened flow signal was
noted across the left internal jugular vein suspicious
of venous thrombosis (Fig 3). This was subsequently
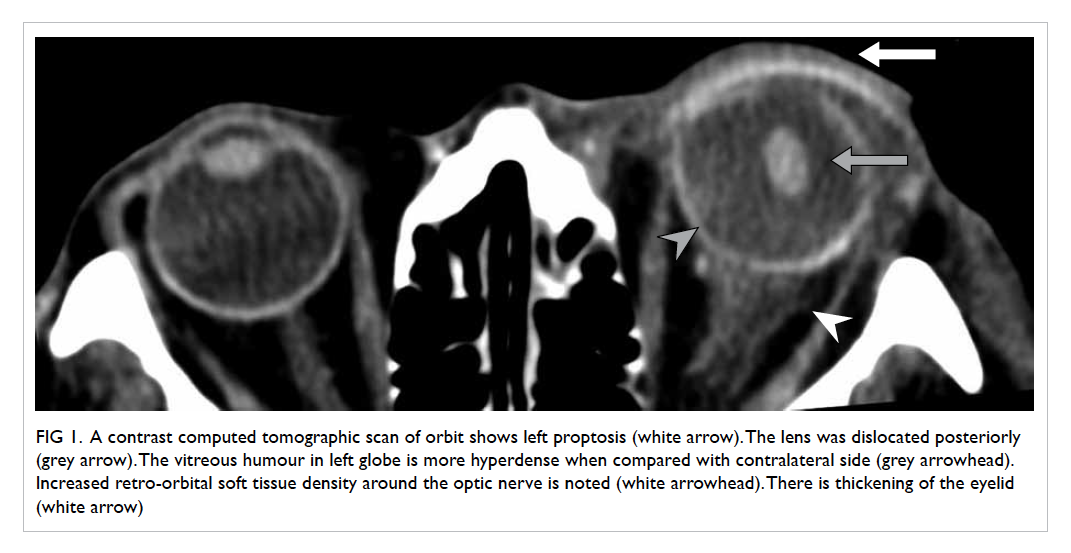
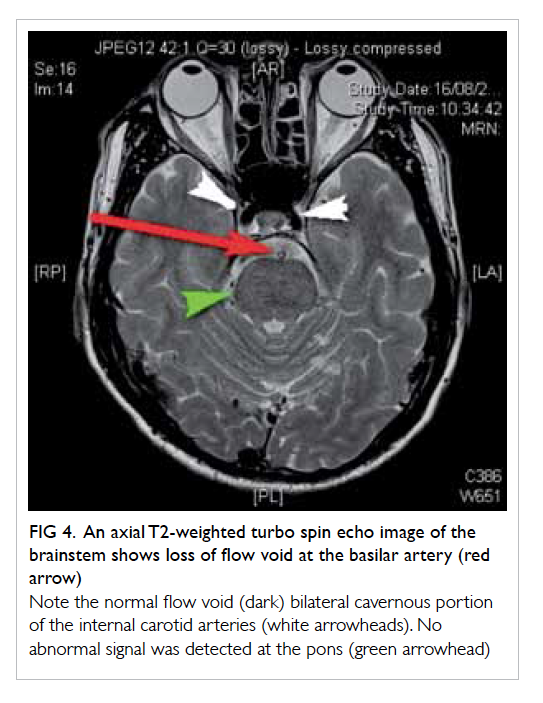
confirmed on contrast CT neck and thorax (Fig 4).
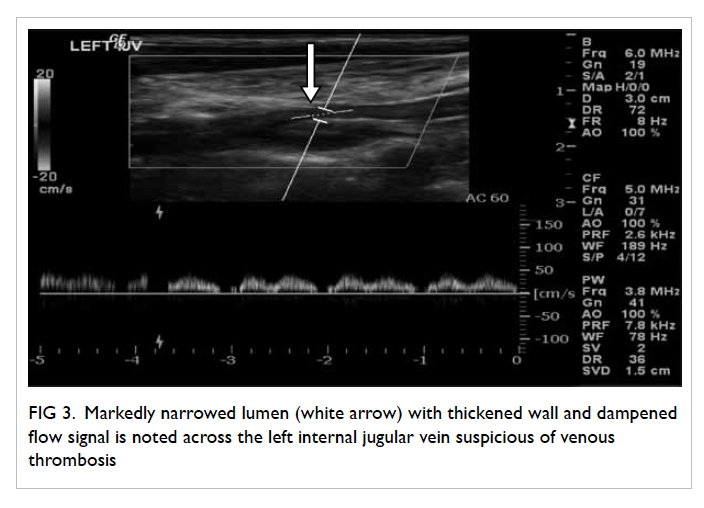
Figure 3. Markedly narrowed lumen (white arrow) with thickened wall and dampened flow signal is noted across the left internal jugular vein suspicious of venous thrombosis
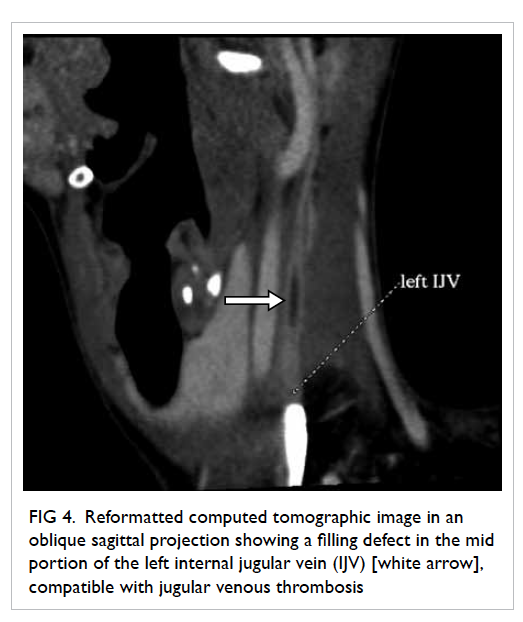
Figure 4. Reformatted computed tomographic image in an oblique sagittal projection showing a filling defect in the mid portion of the left internal jugular vein (IJV) [white arrow], compatible with jugular venous thrombosis
Mycoplasma-Fusobacterium polymerase chain
reaction study confirmed the causative organism as
Fusobacterium necrophorum, which is the classic
bacteria described in Lemierre’s syndrome.
The patient was promptly treated with
intravenous antibiotics and anticoagulant. Her
clinical condition gradually improved and follow-up
CT scan demonstrated interval resolution of
the cavitary lung lesions. She was subsequently
discharged with oral antibiotics and anticoagulants.
Lemierre’s syndrome remains a rare yet
potentially life-threatening disease especially in young
adults.1 A high index of clinical suspicion is therefore
imperative to ensure prompt and timely imaging
investigations that are integral to its diagnosis.
Unrecognised and untreated systemic dissemination
can result in a poor prognosis.2 Contrast-enhanced
CT played a pivotal role in terms of evaluation of the
pulmonary sepsis and assessment of the jugular vein
thrombosis in this patient.3
References
1. Shook J, Trigger C. Lemierre’s Syndrome. West J Emerg
Med 2014;15:125-6. Crossref
2. Lai C, Vummidi DR. Images in clinical medicine.
Lemierre’s Syndrome. N Engl J Med 2004;350:e14. Crossref
3. Screaton NJ, Ravenel JG, Lehner PJ, Heitzman ER, Flower
CD. Lemierre syndrome: forgotten but not extinct—report
of four cases. Radiology 1999;213:369-74. Crossref