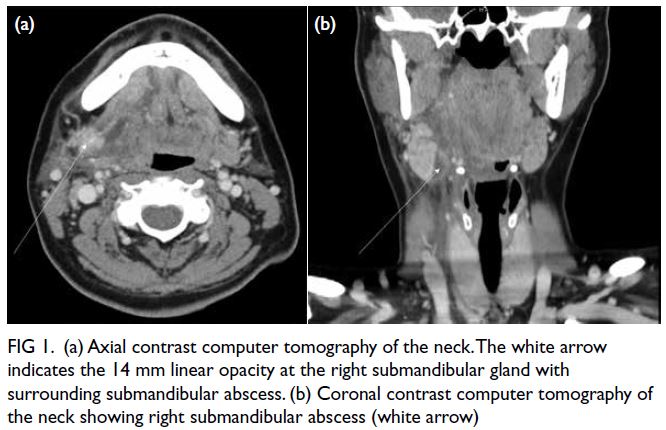
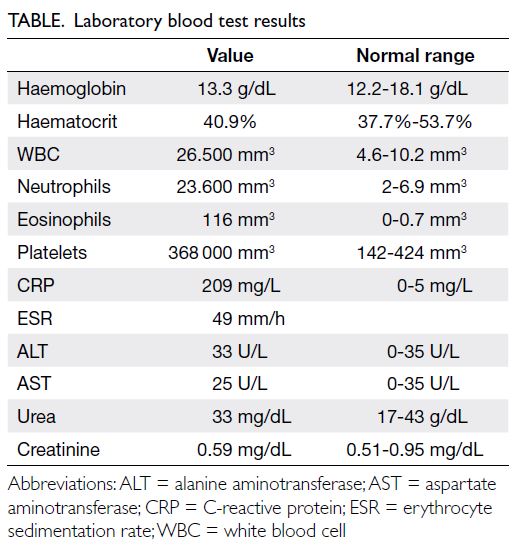
Lipoprotein-X hyperlipidaemia in Chinese paediatric patients with liver graft-versushost disease post-haematopoietic stem cell transplantation: two case reports
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Lipoprotein-X hyperlipidaemia in Chinese
paediatric patients with liver graft-versus-host disease post-haematopoietic stem cell transplantation: two case reports
Wilson YK Chan, MB, BS, MPH1; Eric CY Law, MB, ChB2; TK Ling, MB, BS2; Felix CK Wong, MB, BS, MResMed2; Daniel KL Cheuk, MB, BS, MPH1; Joanna YL Tung, MB, BS, MPH1
1 Department of Paediatrics and Adolescent Medicine, Hong Kong Children’s Hospital, Hong Kong
2 Department of Pathology, Queen Mary Hospital, Hong Kong
Corresponding author: Dr Wilson YK Chan (wykchan@hku.hk)
 Full paper in PDF
Full paper in PDF
Case report
Case 1
An 8-year-old girl with severe aplastic anaemia
and failed immunosuppressive therapy underwent
a matched-sibling bone marrow transplantation.
Neutrophils and platelets were engrafted 23
and 27 days after transplantation, respectively.
Regeneration of marrow and full donor chimerism
were demonstrated 30 days after transplantation.
Progressive liver dysfunction and cholestasis
were noted 5 weeks post-transplantation with
peak alanine aminotransferase level of 878 IU/L
(reference value, <35), aspartate aminotransferase
level 966 IU/L (reference range, 10-40), gamma-glutamyltransferase
level 2065 IU/L (reference
range, 13-28), total bilirubin level 445 μmol/L
(reference range, 10-24), and direct bilirubin level
430 μmol/L (reference range, 5-10). Cholesterol
levels were also elevated with total cholesterol level
of 23.1 mmol/L (reference value, <5.2), high-density
lipoprotein-cholesterol (HDL-C) level 0.5 mmol/L
(reference value, >1.6), and high triglycerides (TG)
level 11.8 mmol/L (reference value, <1.7). Low-density
lipoprotein cholesterol (LDL-C) level could
not be calculated based on indirect quantitation with
the Friedewald equation as per usual practice since
TG level was >4.5 mmol/L; hence, it was measured
directly and was normal at 0.2 mmol/L (reference
value, <4.1).
Apolipoprotein (Apo) A1 level was low at
0.38 g/L (reference range, 1.2-2.0) and Apo B
level was elevated to 1.88 g/L (reference range,
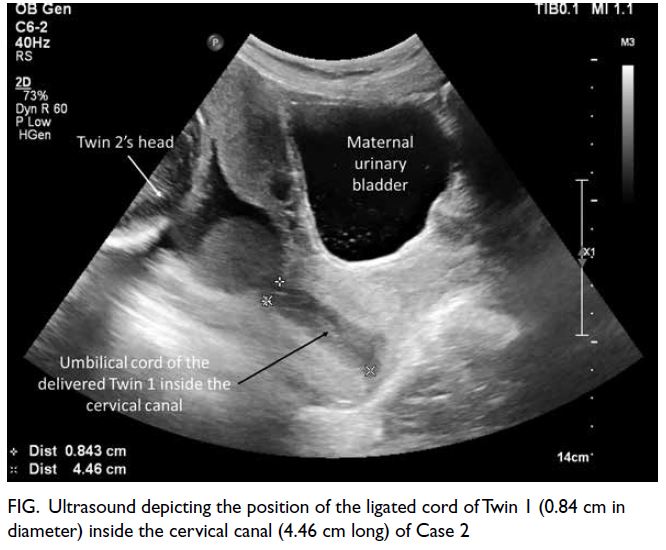
0.41-1.07). Lipoprotein electrophoresis showed
chylomicron and very low–density lipoprotein
bands, and an additional beta-lipoprotein band that
had migrated to cathode was detected, compatible
with lipoprotein-X (Lp-X) [Fig a]. There was no
family history of hypercholesterolaemia and clinical
examination did not reveal any xanthoma. With
improvement of cholestasis, dyslipidaemia gradually resolved by 2 years post-transplant with expectant
management (Fig b and Table).
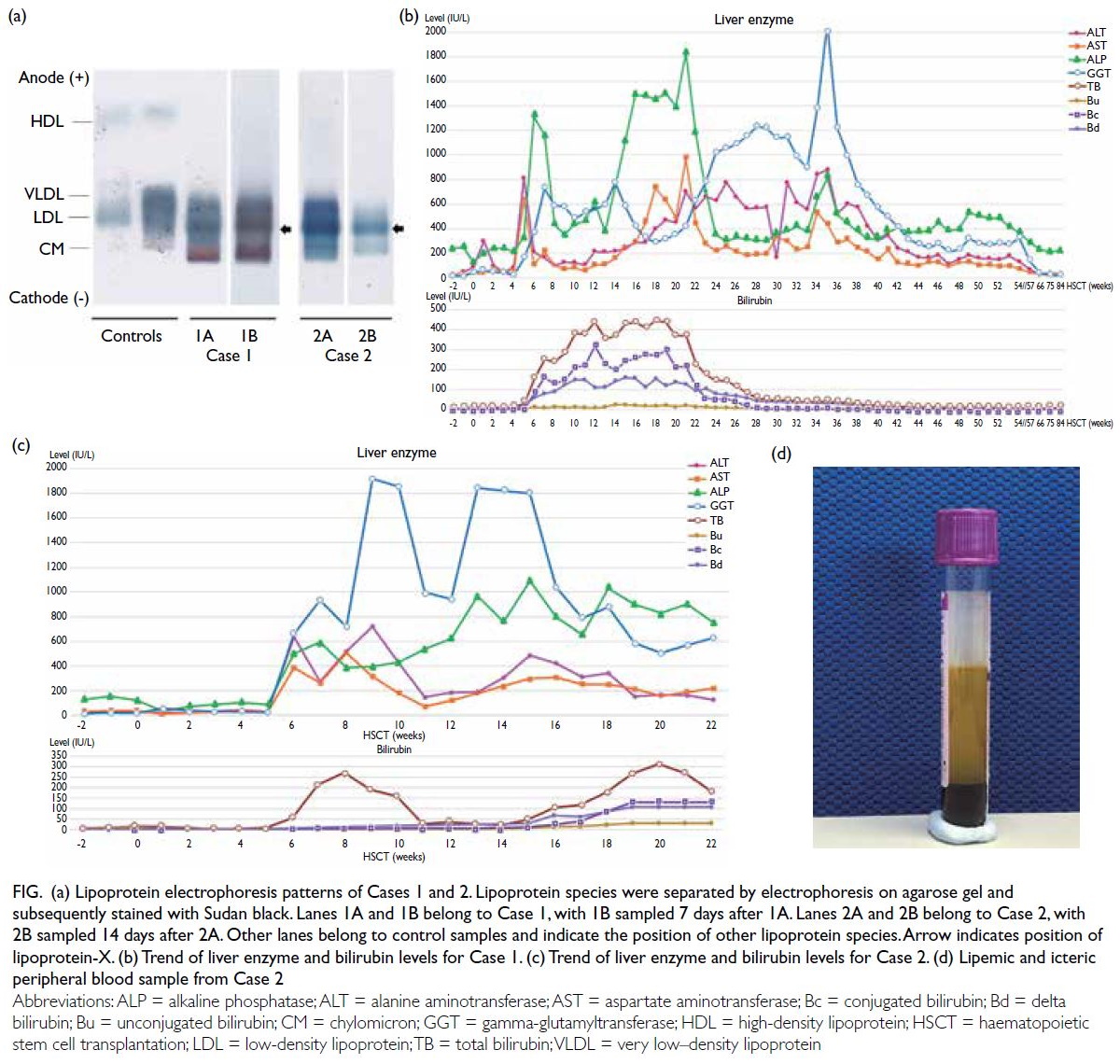
Figure. (a) Lipoprotein electrophoresis patterns of Cases 1 and 2. Lipoprotein species were separated by electrophoresis on agarose gel and subsequently stained with Sudan black. Lanes 1A and 1B belong to Case 1, with 1B sampled 7 days after 1A. Lanes 2A and 2B belong to Case 2, with 2B sampled 14 days after 2A. Other lanes belong to control samples and indicate the position of other lipoprotein species. Arrow indicates position of lipoprotein-X. (b) Trend of liver enzyme and bilirubin levels for Case 1. (c) Trend of liver enzyme and bilirubin levels for Case 2. (d) Lipemic and icteric peripheral blood sample from Case 2
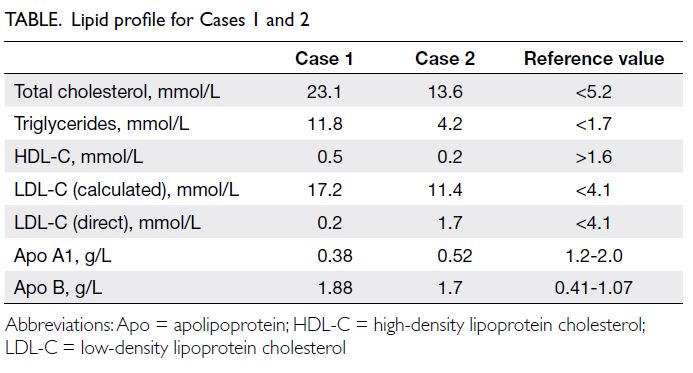
Table. Lipid profile for Cases 1 and 2
Case 2
A 13-year-old boy with stage 4 right adrenal
neuroblastoma and multiple nodal, bone and bone
marrow metastases underwent chemotherapy
(HKPHOSG-NB-07 N7 protocol), gross total tumour
resection, and autologous cord blood transplantation
followed by immunotherapy. Complete remission
was achieved but he had spinal relapse 4.5 years
later presenting with cord compression at the
level of T5-T9 vertebrae warranting emergency
laminectomy and spinal tumour excision. He then
received one cycle of temozolomide and irinotecan
followed by adjuvant radiotherapy 30 Gy/10 Fr to
T5-T9 vertebrae and a haploidentical transplant
with maternal TCRαβ-depleted and CD45RA-depleted
grafts. Total lymphoid irradiation at 8 Gy,
fludarabine 150 mg/m2, thiotepa 10 mg/kg
and melphalan 140 mg/m2 were prescribed as
conditioning. Neutrophils and platelets were
engrafted 10 and 11 days after transplantation,
respectively, with 99% donor chimerism evident
in marrow 30 days after transplantation. His post-transplant
course was complicated by severe
acute graft-versus-host disease (GVHD) involving
skin (grades 2-3), liver (grade 2, biopsy-proven)
and gut (grade 4) requiring prolonged and heavy
immunosuppression including prednisolone,
cyclosporine, and mycophenolate mofetil. He also
had disseminated nocardiosis complicated by left
lower lobe necrotising pneumonia, parapneumonic
effusion, hydropneumothorax and bronchopleural
fistula. Prolonged courses of antimicrobials were
required and included meropenem, levofloxacin,
ceftriaxone, cotrimoxazole, linezolid and
amikacin. Due to liver GVHD, the patient had
grossly deranged liver function 8 months post-transplantation
with peak alanine aminotransferase level of 720 IU/L, aspartate aminotransferase level
506 IU/L, and gamma-glutamyltransferase level
1916 IU/L. His worst cholestasis occurred 2 years
post-transplantation with total bilirubin level of
312 μmol/L and direct bilirubin level 270 μmol/L
(Fig c and Table). He was noted to have a lipemic
blood sample (Fig d) at this time and lipid profile
revealed total cholesterol level of 13.6 mmol/L,
LDL-C (calculated) level 11.4 mmol/L, HDL-C level
0.2 mmol/L and TG level 4.2 mmol/L (Table).
Physical examination did not show any xanthoma.
With the clinical context of severe cholestasis, LDL-C
was measured directly and revealed discordance between the measured and the calculated value
(1.7 mmol/L vs 11.4 mmol/L). Low Apo A1 (0.52 g/L)
and elevated Apo B (1.7 g/L) levels were revealed.
Trace chylomicron band, Lp-X band and faint
lipoprotein Y bands were detected on lipoprotein
electrophoresis (Fig d).
Discussion
Liver GVHD is a known complication of allogeneic
haematopoietic stem cell transplantation. It is
characterised by elevation of hepatic enzymes,
cholestasis, severe hypercholesterolaemia and
hypertriglyceridaemia (in excess of 1000 mg/dL). In
contrast to drug-mediated hypercholesterolaemia
(cyclosporine, sirolimus, mycophenolate and
glucocorticoids) when cholesterol level is usually
<7.8 mmol/L (300 mg/dL) and mediated by LDL-C,
hypercholesterolaemia caused by liver GVHD is
mediated by cholestasis. In general, there are two
main sources that contribute to the liver cholesterol
pool, namely de novo (endogenous) cholesterol
that is mainly synthesised in the liver, and dietary
cholesterol (exogenous). Liver is the primary site of
cholesterol biosynthesis and storage. It is also the
principal site of sterol elimination by converting
cholesterol to bile acids and removing free
cholesterol as neutral sterols via biliary excretion.1 2
In liver GVHD-related cholestasis, impaired bile
flow results in accumulation of cholesterol and bile
salts, and hence elevated LDL-C level. Lipoprotein-X
is another major cause of hyperlipidaemia in
cholestasis when bile constituents reflux from
the bile ducts or hepatocytes to the blood stream.
Lipoprotein-X particles are formed when bile
lipoprotein enters the blood stream and incorporates
TG, Apo C and esterified cholesterol. Unlike LDL-C,
Lp-X does not contain Apo B, the most important
ligand to the hepatic LDL-C receptor. Therefore,
Lp-X cannot be internalised into hepatocytes.
Since Lp-X hypercholesterolaemia is not due to
overproduction by hepatocytes, use of medications
such as statins to downregulate cholesterol synthesis
is ineffective.3 In addition, since Lp-X does not
contain Apo B, which is the major component of
LDL and one of the most important factors in the
pathogenesis of atherosclerotic plaques, it is not
atherogenic.4 Neither of our cases reported here
had any complications of hypercholesterolaemia
including exanthemata, retinal thromboembolism
and pulmonary cholesteroloma. Nonetheless Lp-X
may be associated with hyperviscosity syndrome
and plasma exchange or apheresis may be indicated.5
As reported in the literature,
hypercholesterolaemia secondary to intrahepatic
cholestasis caused by liver GVHD can appear
at any time between 2 months and 2 years after
haematopoietic stem cell transplantation. The condition can be easily diagnosed by demonstrating
discordance between calculated and directly
measured LDL-C level, as well as lipoprotein
electrophoresis. With the resolution of cholestasis,
Lp-X will resolve with no specific treatment.
To conclude, severe hypercholesterolaemia
mediated by Lp-X in post-haematopoietic stem
cell transplantation patients with liver GVHD is a
recognised yet overlooked phenomenon. Reports in
the literature are limited for both adult and paediatric
populations. To the best of our knowledge, this
is the first report in Chinese children. Transplant
physicians and endocrinologists should have an
increased awareness of this association and avoid
unnecessary and ineffective use of statins.
Author contributions
Concept or design: WYK Chan, JYL Tung.
Acquisition of data: WYK Chan, ECY Law, TK Ling, FCK Wong, JYL Tung.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: WYK Chan.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: WYK Chan, ECY Law, TK Ling, FCK Wong, JYL Tung.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: WYK Chan.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
Patients were treated in accordance with the Declaration of Helsinki. Consents for treatment, procedures and publication were obtained.
References
1. Fellin R, Manzato E. Lipoprotein-X fifty years after its
original discovery. Nutr Metab Cardiovasc Dis 2019;29:4-8. Crossref
2. Nemes K, Åberg F, Gylling H, Isoniemi H. Cholesterol
metabolism in cholestatic liver disease and liver
transplantation: from molecular mechanisms to clinical
implications. World J Hepatol 2016;8:924-32. Crossref
3. Zidan H, Lo S, Wiebe D, Talano J, Alemzadeh R. Severe
hypercholesterolemia mediated by lipoprotein X in a
pediatric patient with chronic graft-versus-host disease of
the liver. Pediatr Blood Cancer 2008;50:1280-1. Crossref
4. Miida T, Hirayama S. Controversy over the atherogenicity of lipoprotein-X. Curr Opin Endocrinol Diabetes Obes
2019;26:117-23. Crossref
5. Heinl RE, Tennant HM, Ricketts JC, et al. Lipoprotein-X disease in the setting of severe cholestatic hepatobiliary autoimmune disease. J Clin Lipidol 2017;11:282-6. Crossref