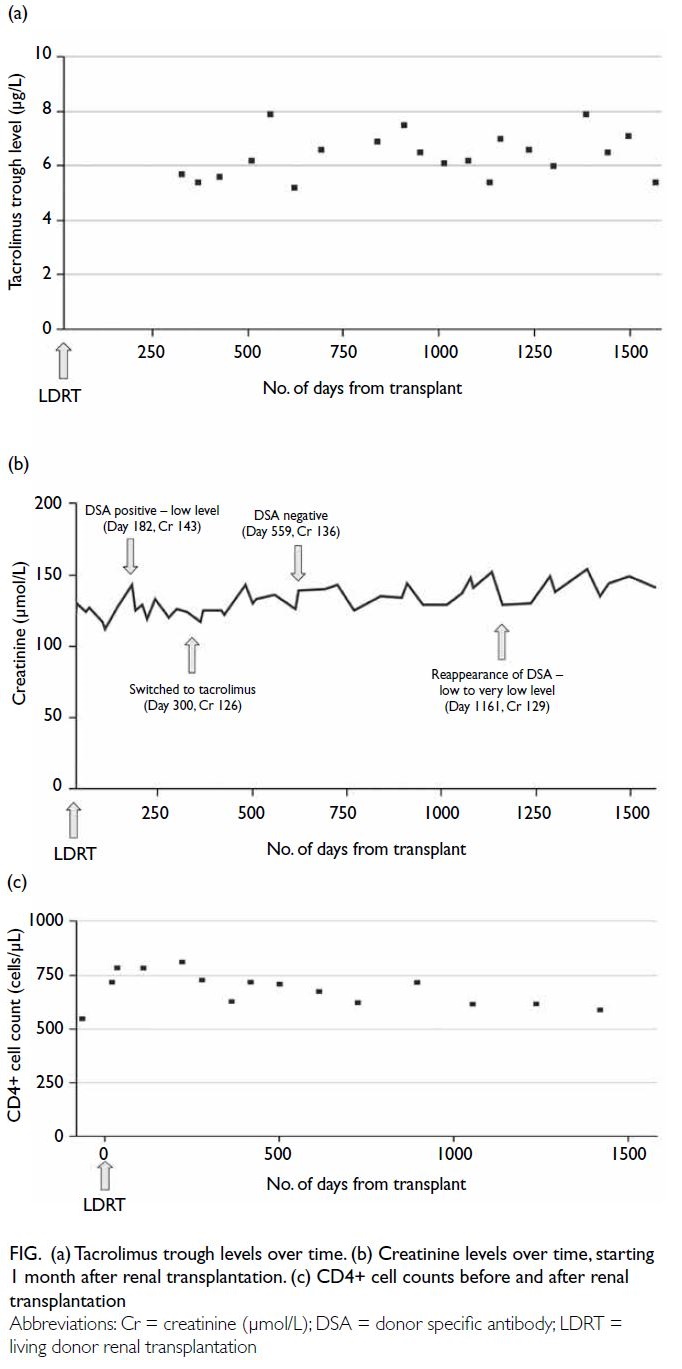
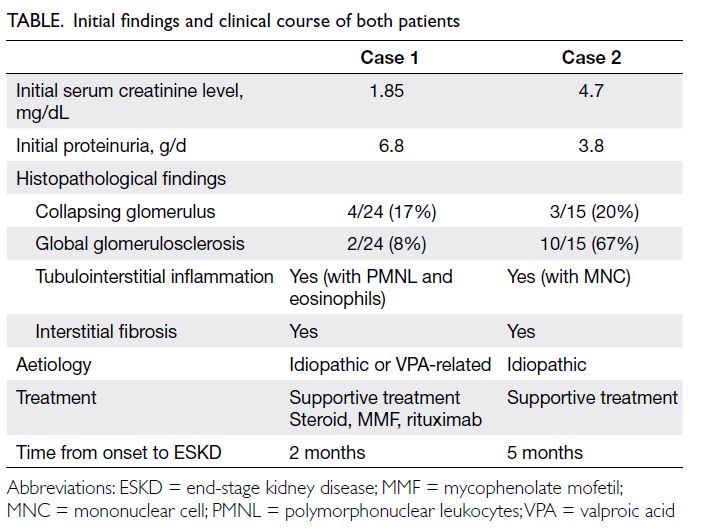
Rethink personalised sudden cardiac death risk assessment in non-dilated left ventricular cardiomyopathy: a case report
Hong Kong Med J 2025 Jun;31(3):236–9 | Epub 14 Apr 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Rethink personalised sudden cardiac death risk assessment in non-dilated left ventricular cardiomyopathy: a case report
Kevin WC Lun, MB, BS, MRCP1 #; Jonan CY Lee, MB, ChB, FRCR2; Eric CY Wong, MB, BS, FHKCP1; Michael KY Lee, MB, BS, FHKCP1; Derek PH Lee, MB, ChB, FHKCP1 #
1 Division of Cardiology, Department of Medicine, Queen Elizabeth Hospital, Hong Kong SAR, China
2 Department of Diagnostic and Interventional Radiology, Queen Elizabeth Hospital, Hong Kong SAR, China
# Equal contribution
Corresponding author: Dr Kevin WC Lun (lkw708@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Case presentation
A 56-year-old man was presented to the emergency
department of our institution in June 2023 and has
had been followed up in our medical clinic for 1 year
prior to his current hospital admission. He had been
diagnosed with frequent symptomatic premature
ventricular complexes with an ectopic burden of
8.2% on extended ambulatory rhythm monitoring.
There were also multiple recorded episodes of non-sustained
ventricular tachycardia. Beta-blocker
was initiated and uptitrated according to clinical
symptoms. His family history was remarkable for
the sudden cardiac death of his father at the age of
64 years. Subsequent transthoracic echocardiogram
of the patient revealed a global hypokinetic left
ventricle with a biplane-measured left ventricular
ejection fraction (LVEF) of 45%. There was mild
left atrial enlargement with a two-dimensional area
of 25.7 cm2 but no other structural abnormalities.
Computed tomography coronary angiogram showed
mild to moderate coronary artery disease in three
vessels and guideline-directed medical treatment
was initiated. Cardiac magnetic resonance imaging
(CMR) was scheduled to assess cardiac structures
and function, as well as tissue characterisation for
features of non-ischaemic cardiomyopathy. He
had been scheduled to undergo catheter ablation
for frequent symptomatic premature ventricular
complexes.
The patient presented to our emergency
department with out-of-hospital cardiac arrest. He
had been found collapsed adjacent to a swimming
pool and a bystander had initiated cardiopulmonary
resuscitation. Spontaneous circulation was restored
shortly after a single defibrillation delivered by
an automated external defibrillator and he was
transferred to our hospital immediately. On arrival at
the emergency department, he was hemodynamically
stable with a Glasgow Coma Scale score of 15.
High-sensitive troponin I level was elevated
at 44.9 ng/L. Electrocardiogram showed sinus
rhythm of 71 beats/min with occasional premature ventricular complexes. There was no ST-segment
elevation or significant conduction abnormalities.
Bedside echocardiogram showed a similar biplane-measured
LVEF of 43% with global hypokinesia.
Urgent coronary angiogram showed non-occlusive
moderate to severe coronary artery disease in three
vessels. Given his current presentation, together
with angiographic progression in coronary artery
disease, complete revascularisation was performed
uneventfully. He was then transferred to our cardiac
care unit postoperatively for close monitoring.
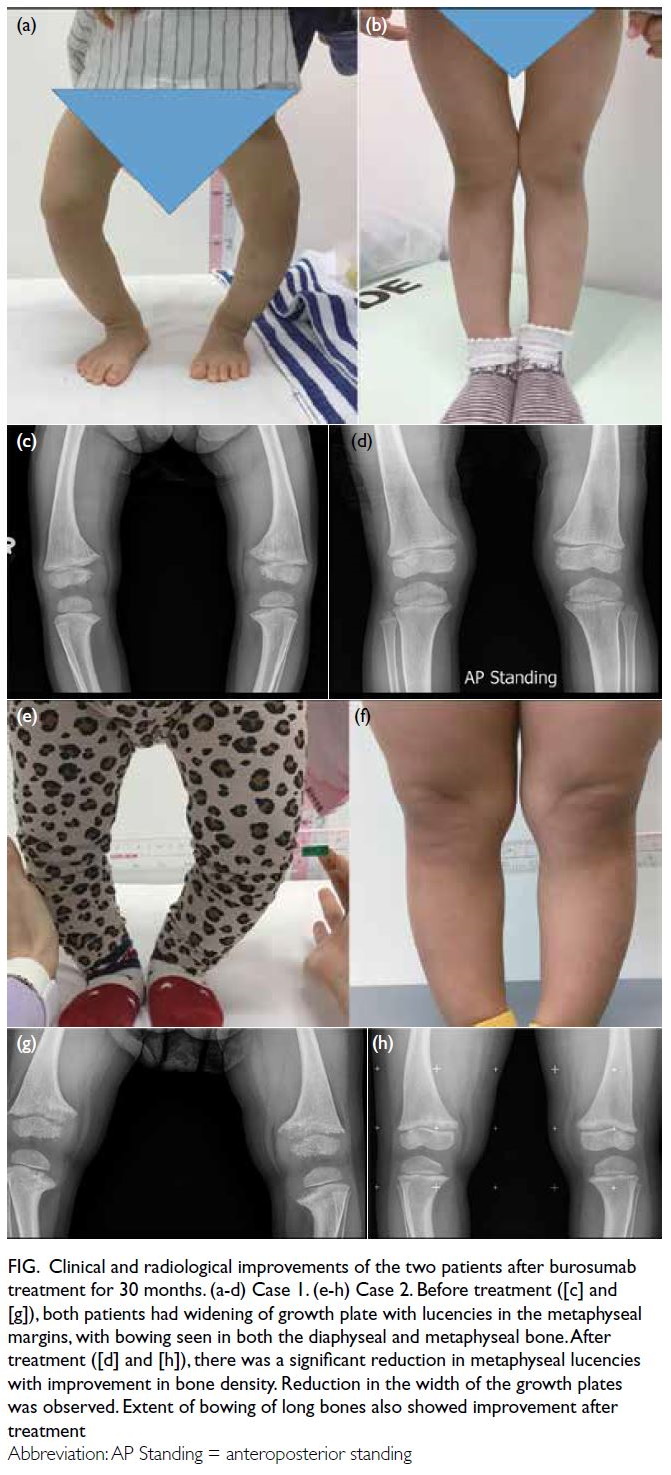
Inpatient CMR revealed a non-dilated left ventricle
with mildly reduced LVEF of 43% with global
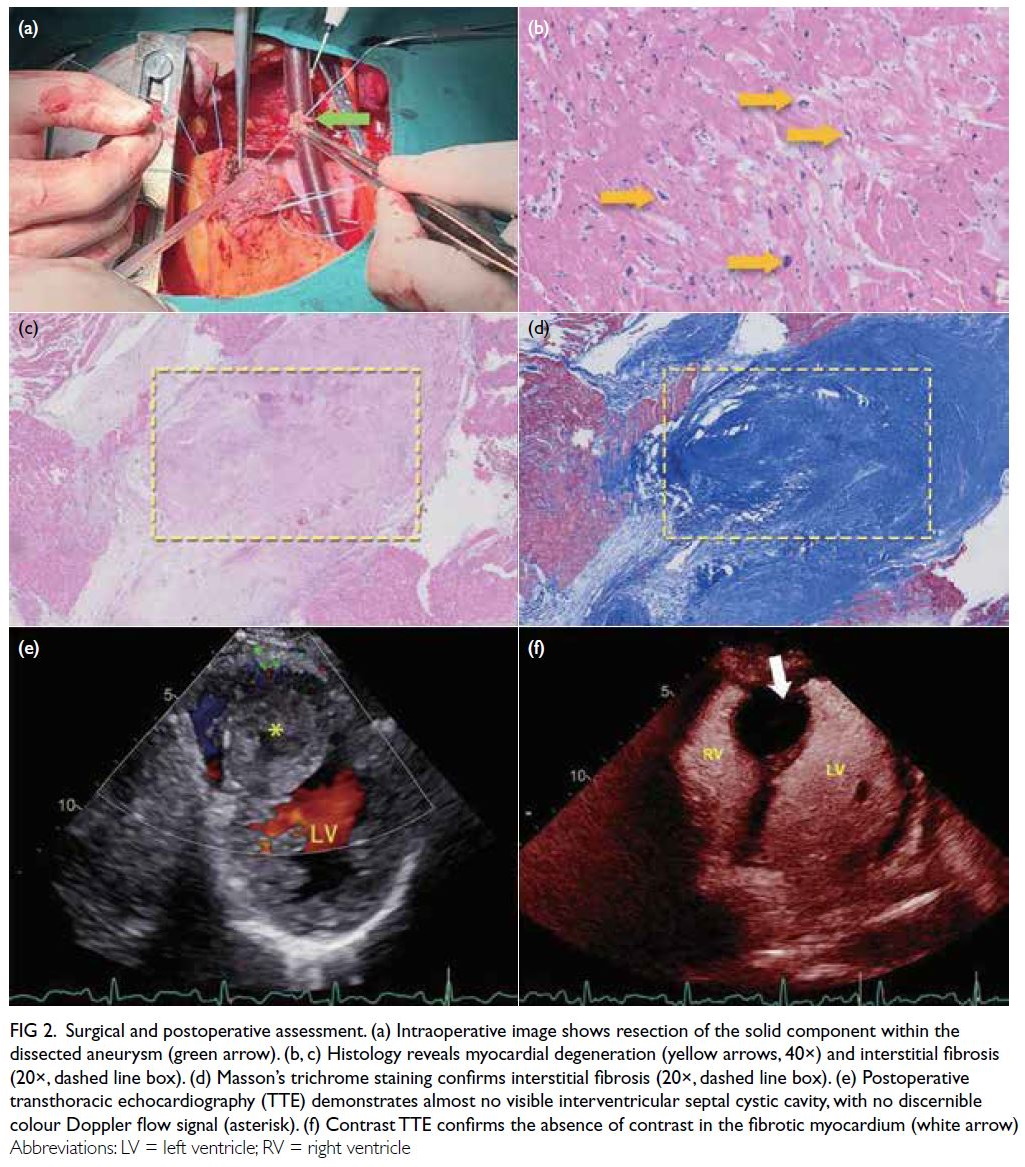
hypokinesia. There was multifocal patchy mid-wall
and subepicardial late gadolinium enhancement
(LGE) at the mid-ventricular anterior, anteroseptal
and anterolateral walls, as well as basal to mid-ventricular
inferior and inferolateral walls (Fig 1). There was no evidence of myocardial infarct.
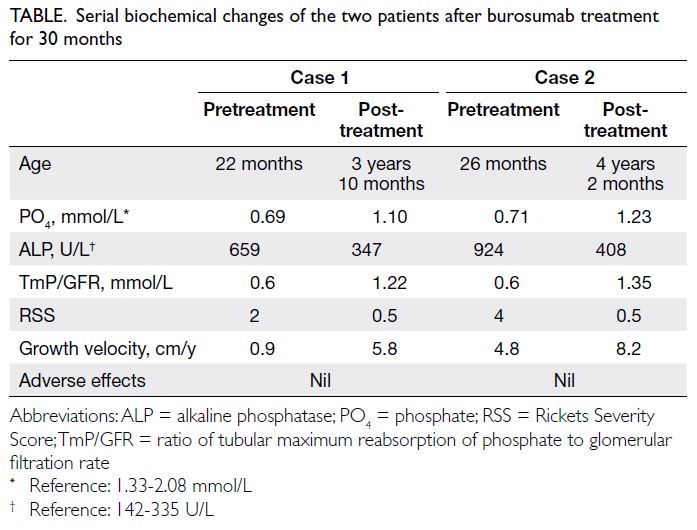
Parametric mapping showed a mild increase in
myocardial T1 with values up to 1067 ms to 1080
ms (native T1 values in healthy subjects obtained in
our Aera 1.5T magnetic resonance imaging scanner
[Siemens, Munich, Germany] is 996±26 ms for
males), suggestive of mild diffuse interstitial fibrosis
(Fig 2). Prior to hospital discharge, a transvenous
implantable cardioverter defibrillator (ICD) was
implanted for secondary prevention. Subsequent
genetic testing identified a heterozygous pathogenic
truncating variant NM_001458.5(FLNC):c.3279del
p.(Gly1094Alafs*4) in the filamin-C gene. The
final clinical diagnosis was sudden cardiac arrest
secondary to filamin-C variant–associated
cardiomyopathy in a patient with non-dilated left
ventricular cardiomyopathy (NDLVC) and mid-range
ejection fraction.
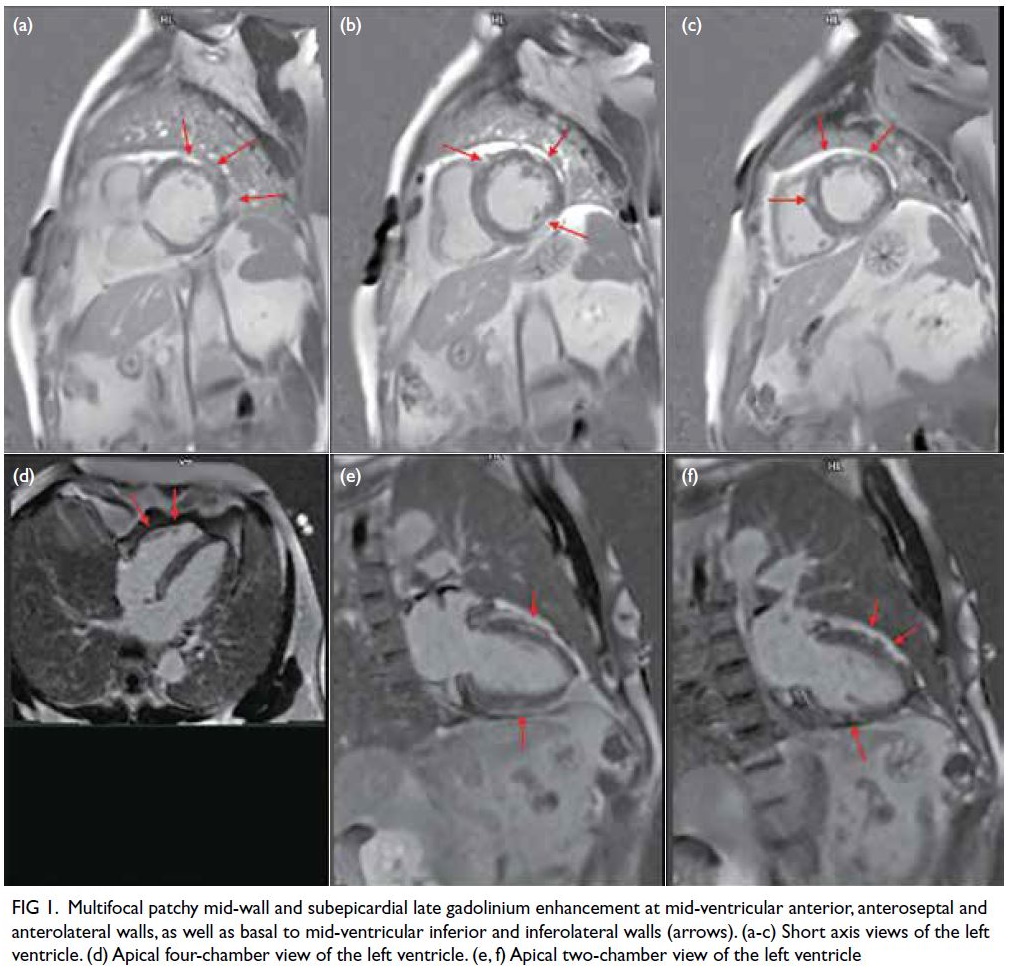
Figure 1. Multifocal patchy mid-wall and subepicardial late gadolinium enhancement at mid-ventricular anterior, anteroseptal and anterolateral walls, as well as basal to mid-ventricular inferior and inferolateral walls (arrows). (a-c) Short axis views of the left ventricle. (d) Apical four-chamber view of the left ventricle. (e, f) Apical two-chamber view of the left ventricle
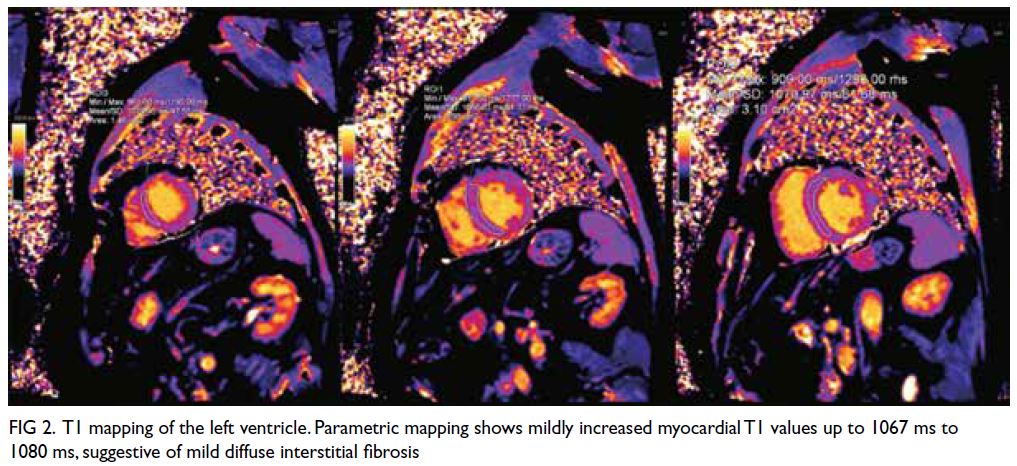
Figure 2. T1 mapping of the left ventricle. Parametric mapping shows mildly increased myocardial T1 values up to 1067 ms to 1080 ms, suggestive of mild diffuse interstitial fibrosis
Discussion
The filamin-C gene encodes the filamin-C protein
that plays essential roles in the sarcomere stability
in cardiac muscles. Filamin-C variants have been
increasingly recognised as an important cause of cardiomyopathy. It has been identified in
approximately 3% to 4% of patients with dilated
cardiomyopathy and commonly presents in early-to-mid
adulthood with high arrhythmic risks.1
According to the 2021 European Society of
Cardiology (ESC) Guidelines for the diagnosis
and treatment of acute and chronic heart failure,2
primary prevention ICD is indicated in patients
with symptomatic heart failure with an LVEF of lower than 35%
despite optimal medical treatment and a reasonable
quality of life. Such recommendation is supported
by numerous landmark trials including MADIT
(Multicenter Automatic Defibrillator Implantation
Trial),3 DEFINITE (DEFibrillators In Non-Ischemic
Cardiomyopathy Treatment Evaluation),4 and SCD-HeFT
(the Sudden Cardiac Death in Heart Failure
Trial).5 In addition, several additional clinical
risk factors should also be considered in sudden
cardiac death risk assessment, especially in patients
with non-ischaemic cardiomyopathy.2 These risk
factors include significant LGE on CMR, younger
age, and specific genotypes. Nonetheless these
recommendations are ambiguous, and the guideline
has not defined, for example, the burden of LGE
and variant mechanisms in several high-risk genes
that would warrant ICD implantation. Moreover,
there are limited recommendations for primary
prevention ICD in patients with heart failure with
mid-range or preserved ejection fraction.
In the updated 2023 ESC Guidelines for
the management of cardiomyopathies,6 a new
entity of NDLVC is introduced. An algorithm for
consideration of primary prevention ICD similar
to that for patients with dilated cardiomyopathy is
recommended in this patient population. Patient
genotype and imaging features on CMR have been
proposed in the early sudden cardiac death risk
assessment for patients with NDLVC. A previous
study has demonstrated a higher rate of malignant
arrhythmic events in patients who are genotype-positive,
compared with their genotype-negative
counterparts.7 Such association has been observed
irrespective of LVEF. Variants in certain genes
including lamin A/C, phospholamban, filamin-C,
RNA-binding motif protein 20, desmoplakin and
plakophilin-2 are associated with a high risk of
malignant ventricular arrhythmias and sudden
cardiac death. Apart from genotype information,
the presence and distribution of LGE on CMR,
such as a ring-like pattern of LGE, has also been
shown to be a strong risk marker for ventricular
arrhythmias.8 Hence, based on the current ESC
Guidelines,6 primary prevention ICD should be
considered in patients with NDLVC and high-risk
genotype and in the presence of additional
risk factors such as syncope and LGE on CMR,
irrespective of LVEF.
Our case highlights the need to incorporate a patient’s genotype and imaging features on CMR into
the personalised risk assessment for sudden cardiac
death in patients with NDLVC. This will facilitate
a more comprehensive and informative discussion
about the indication for primary prevention ICD
and improve the clinical outcome for patients with
cardiomyopathy.
Author contributions
Concept or design: KWC Lun, DPH Lee.
Acquisition of data: All authors.
Analysis or interpretation of data: KWC Lun, DPH Lee.
Drafting of the manuscript: KWC Lun, DPH Lee.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: All authors.
Analysis or interpretation of data: KWC Lun, DPH Lee.
Drafting of the manuscript: KWC Lun, DPH Lee.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study received no specific grant from any funding agency
in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of
Helsinki. The patient provided consent for all treatments and
procedures, and consent for publication of the case report.
References
1. Agarwal R, Paulo JA, Toepfer CN, et al. Filamin C
cardiomyopathy variants cause protein and lysosome
accumulation. Circ Res 2021;129:751-66. Crossref
2. McDonagh TA, Metra M, Adamo M, et al. 2021 ESC
Guidelines for the diagnosis and treatment of acute and
chronic heart failure. Eur Heart J 2021;42:3599-726. Crossref
3. Moss AJ, Zareba W, Hall WJ, et al. Prophylactic
implantation of a defibrillator in patients with myocardial
infarction and reduced ejection fraction. N Engl J Med
2002;346:877-83. Crossref
4. Kadish A, Dyer A, Daubert JP, et al. Prophylactic
defibrillator implantation in patients with nonischemic
dilated cardiomyopathy. N Engl J Med 2004;350:2151-8. Crossref
5. Bardy GH, Lee KL, Mark DB, et al. Amiodarone or an
implantable cardioverter-defibrillator for congestive heart
failure. N Engl J Med 2005;352:225-37. Crossref
6. Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC
Guidelines for the management of cardiomyopathies. Eur
Heart J 2023;44:3503-626. Crossref
7. Escobar-Lopez L, Ochoa JP, Mirelis JG, et al. Association
of genetic variants with outcomes in patients with
nonischemic dilated cardiomyopathy. J Am Coll Cardiol
2021;78:1682-99. Crossref
8. Meier C, Eisenblätter M, Gielen S. Myocardial late
gadolinium enhancement (LGE) in cardiac magnetic
resonance imaging (CMR)—an important risk marker for
cardiac disease. J Cardiovasc Dev Dis 2024;11:40. Crossref