SARS-CoV-2–associated myopathy with positive anti–Mi-2 antibodies: a case report
Hong Kong Med J 2023 Apr;29(2):170–2 | Epub 17 Mar 2023
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
SARS-CoV-2–associated myopathy with positive anti–Mi-2 antibodies: a case report
Aleksandra Plavsic, MD1,2; Snezana Arandjelovic, MD, PhD1,2; Aleksandra Peric Popadic, MD, PhD1,2; Jasna Bolpacic, MD, PhD1,2; Sanvila Raskovic, MD, PhD1,2; Rada Miskovic, MD1,2
1 Clinic of Allergy and Immunology, University Clinical Centre of Serbia, Beograd, Serbia
2 Medical Faculty, University of Belgrade, Beograd, Serbia
Corresponding author: Dr Rada Miskovic (rada_delic@hotmail.com)
 Full paper in PDF
Full paper in PDF
Case report
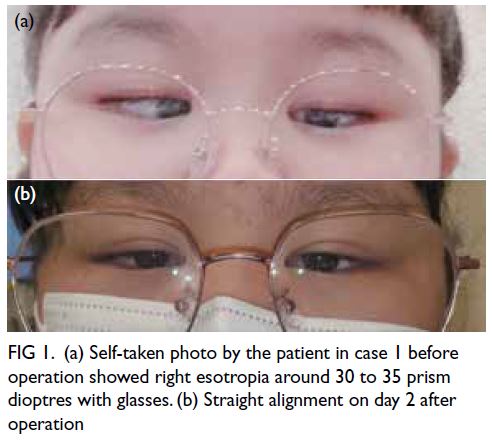
A 36-year-old female presented to the Emergency
Department of the University Clinical Centre of
Serbia in August 2020 with bilateral weakness and
aches in the proximal muscles of the upper and
lower extremities, limited limb movement, and
poor tolerance of physical exertion. Symptoms
had developed 4 weeks after hospitalisation for
coronavirus disease 2019 (COVID-19) pneumonia
and progressed rapidly over the next weeks
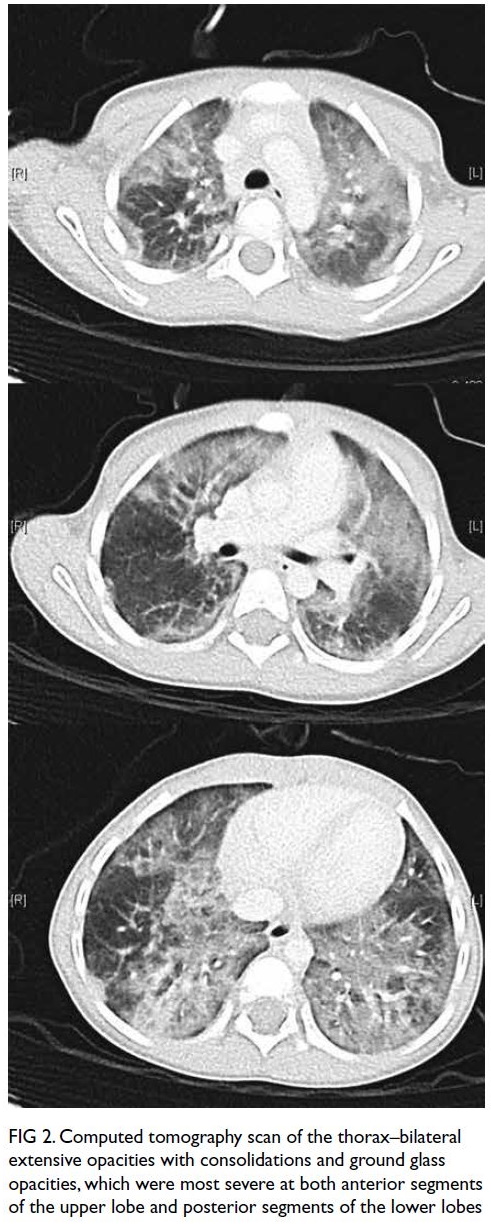
(Fig). During her hospitalisation for COVID-19,
she was treated with the corticosteroid (CS)
methylprednisolone 0.5 mg/kg for 10 days, and
prophylactic low-molecular-weight heparin,
azithromycin (500 mg/day for 3 days), and antipyretics. Physical examination
revealed weakness of the proximal muscles (grade
3/5) but no other abnormalities. Nasal swab
screening for severe acute respiratory syndrome
coronavirus 2 (SARS-CoV-2) by polymerase chain
reaction test was negative. Blood work-up showed elevated levels of creatine kinase (CK) [28 731 U/L],
myoglobin, and troponin (Table). Renal function
tests were normal (blood urea nitrogen: 8.0 mmol/L,
serum creatinine: 45 μmol/L, estimated glomerular
filtration rate: >60 mL/min). Immunoserological
analysis was positive for assessment of antinuclear
antibodies (1:640) and myositis profile (Mi-2++ and Ro-52++). The patient was then referred to the
Clinic of Allergy and Immunology of the same centre
for further evaluation. Electromyoneurography of
the upper and lower extremities revealed moderate-to-severe myopathic lesions in the proximal muscles
with a pattern characteristic of inflammatory
myopathies. Testing of respiratory muscle strength
revealed decreased maximal inspiratory pressure
of 62%. Lung computed tomography scan and
echocardiography were normal. Muscle biopsy and
magnetic resonance imaging were not performed
due to temporary restrictions during the pandemic.
The remainder of a thorough work-up was normal. Autoimmune myopathy associated with COVID-19
was suspected and the patient was prescribed high-dose
CS (1 mg/kg/day) followed by pulse therapy of
500 mg/day methylprednisolone for 3 consecutive
days. Methotrexate (22.5 mg/week) was introduced.
No clinical or laboratory improvement was evident
after 2 weeks, hence intravenous immunoglobulins
(0.4 g/kg/day) were given for 5 days. This therapy
led to significant clinical improvement and a gradual
decline in muscle enzymes. During follow-up, a trial
of CS withdrawal and methotrexate dose reduction
resulted in worsening of proximal muscle weakness
and rise in serum CK. After 1 year of follow-up, the
patient remained on methotrexate 20 mg/week and
prednisone 5 mg/day. Repeated immunoserological
analysis was still positive for antinuclear antibodies
(1:640) and anti–Mi-2 antibodies.
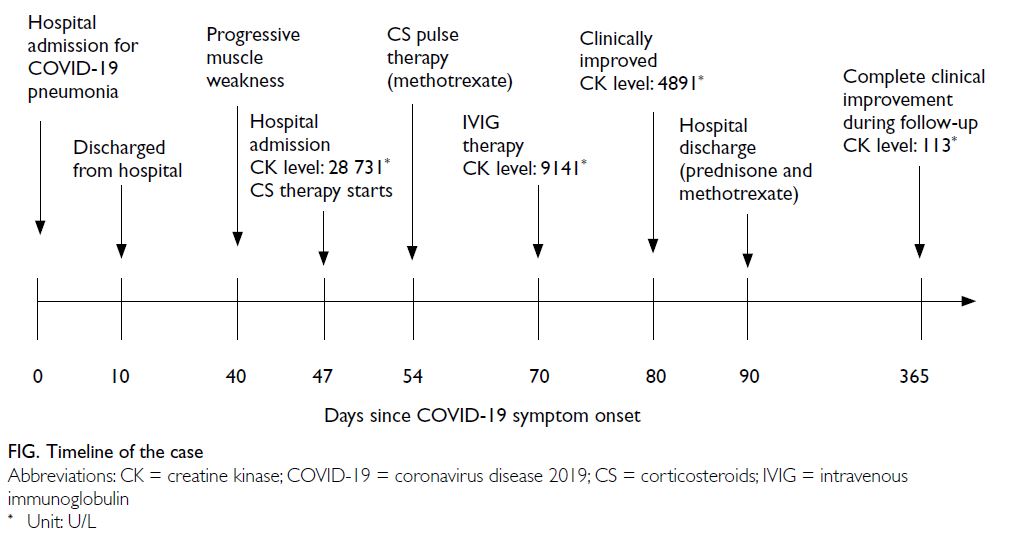
Figure. Timeline of the case
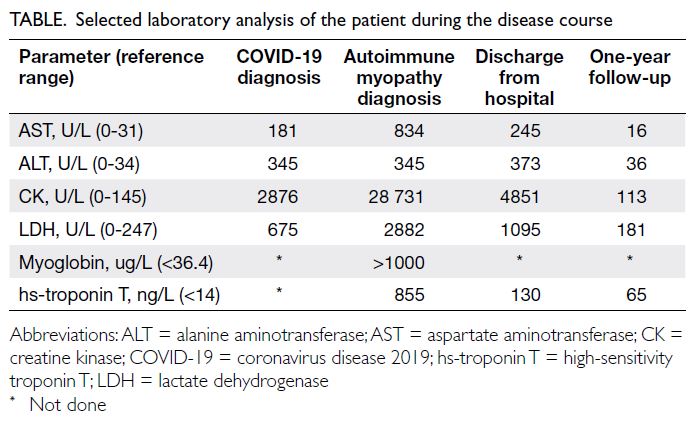
Table. Selected laboratory analysis of the patient during the disease course
Discussion
Myalgias, muscle weakness, and elevation of muscle
enzymes are commonly seen in COVID-19 patients,
but are typically resolved within a few weeks with
conservative treatment. Direct viral invasion of
muscles, toxic effects of cytokines and dysregulated
immune stimulation have been proposed as
possible mechanisms. There are several reports of
COVID-19–related myositis/rhabdomyolysis with
serum CK level as high as 427 656 U/L.1 2 Most
patients recover with conservative treatment. In
our patient, a mild increase in muscle enzymes was
noticed during COVID-19 infection. Nonetheless
early signs of myopathy may have been masked
by the CS therapy prescribed for COVID-19
pneumonia. Due to the presence of anti–Mi-2 and
anti–Ro-52 antibodies, which are characteristic
of dermatomyositis, we carefully examined the
skin and nailfolds, but there were no suggestive
findings at first presentation or subsequent follow-up.
A limitation of our work was the lack of muscle
histopathology or magnetic resonance imaging. Nonetheless the clinical picture, 1-year disease
course, electromyographic pattern typical of
inflammatory myopathies, and persistent positivity
for anti–Mi-2 antibodies suggest that SARS-CoV-2
infection may have triggered the development of
autoimmune myopathy in our patient.
A case of COVID-19–associated inflammatory
myopathy with severe facial, bulbar, proximal limb
weakness, and elevated CK level, suggestive muscle
biopsy and positive anti-SSA, anti–SAE-1, and
anti-Ku antibodies, has been reported. The patient
was successfully treated with a 5-day course of 1000
mg methylprednisolone.3 Nonetheless there are no
data on the subsequent clinical course.
Recently, a COVID-19–associated myopathy
caused by type I interferonopathy has been
described.4 The patient presented with general
weakness, myalgias, fever, bibasilar lung infiltrates,
and significantly elevated serum CK and troponin
levels. Immunohistochemical analysis of deltoid
muscle biopsy specimen revealed abnormal
expression of major histocompatibility complex
class I antigen on sarcolemma and sarcoplasm,
and presence of myxovirus resistance protein A on
muscle fibres and capillaries. The authors speculated
that increased expression of type I interferon was
responsible for SARS-CoV-2 myopathy in their
patient through up-regulation of proteins that are
toxic to muscle cells.4 Nonetheless deposition of
myxovirus resistance protein A in muscle fibres and
capillaries is also an early feature of dermatomyositis.
Given the lack of data on immunoserological
analysis and follow-up of the patient, an early phase
of dermatomyositis cannot be excluded.
The number of reports of autoimmune disease
developing in patients with COVID-19 is increasing.
Molecular mimicry has been proposed as a potential
mechanism.5 A recent study identified three
immunogenic linear epitopes with high sequence
identity to SARS-CoV-2 proteins in patients with
dermatomyositis, implying a possible contribution
of SARS-CoV-2 to the development of autoimmune
inflammatory myopathies.6 Additional mechanisms
may also be involved, highlighting an urgent need
to better understand the immune processes that
underlie viral-induced autoimmunity in COVID-19.
Physicians should carefully evaluate patients
who present with progressive elevation of muscle
enzymes and be alert to the possible occurrence
of autoimmune myopathy triggered by COVID-19. Early diagnosis facilitates timely initiation
of adequate treatment, preventing long-term
consequences and complications.
Author contributions
Concept or design: A Plavsic, S Arandjelovic, A Peric Popadic, S Raskovic, R Miskovic.
Acquisition of data: A Plavsic, J Bolpacic, R Miskovic.
Analysis or interpretation of data: A Plavsic, A Peric Popadic, S Raskovic, J Bolpacic, R Miskovic.
Drafting of the manuscript: A Plavsic, S Arandjelovic, J Bolpacic, R Miskovic.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: A Plavsic, J Bolpacic, R Miskovic.
Analysis or interpretation of data: A Plavsic, A Peric Popadic, S Raskovic, J Bolpacic, R Miskovic.
Drafting of the manuscript: A Plavsic, S Arandjelovic, J Bolpacic, R Miskovic.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Acknowledgement
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
Ethics approval was not required as per guidelines for publishing case reports of Ethics Committee of the University Clinical Centre of Serbia. Patient consent has been obtained
concerning treatment and procedures, and the patient has given written informed consent to the publication.
References
1. Beydon M, Chevalier K, Al Tabaa O, et al. Myositis as a manifestation of SARS-CoV-2. Ann Rheum Dis 2021;80:e42. Crossref
2. Gefen AM, Palumbo N, Nathan SK, Singer PS, Castellanos-Reyes LJ, Sethna CB. Pediatric COVID-19–associated rhabdomyolysis: a case report. Pediatr Nephrol 2020;35:1517-20. Crossref
3. Zhang H, Charmchi Z, Seidman RJ, Anziska Y, Velayudhan V, Perk J. COVID-19–associated myositis with severe proximal and bulbar weakness. Muscle Nerve 2020;62:E57-60. Crossref
4. Manzano GS, Woods JK, Amato AA. Covid-19–associated myopathy caused by type I interferonopathy. N Engl J Med 2020;383:2389-90. Crossref
5. Kanduc D. From anti–SARS-CoV-2 immune responses to COVID-19 via molecular mimicry. Antibodies (Basel) 2020;9:33. Crossref
6. Megremis S, Walker TD, He X, et al. Antibodies against immunogenic epitopes with high sequence identity to SARS-CoV-2 in patients with autoimmune dermatomyositis. Ann Rheum Dis 2020;79:1383-6. Crossref