Purtscher-like retinopathy in a patient with lupus: a case report
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Purtscher-like retinopathy in a patient with lupus: a case report
Cynthia KL Cheng, MB, ChB1; Kenneth KH Lai, MB, ChB1; Andrew KT Kuk, MB, BS, MRCS1; Tracy HT Lai, FCOphthHK, FHKAM (Ophthalmology)2,3; Sarah T Wang, MB, BS, MRCS1; Simon TC Ko, FRCS (Edin), FHKAM (Ophthalmology)1
1 Department of Ophthalmology, Tung Wah Eastern Hospital, Hong Kong
2 Department of Ophthalmology, United Christian Hospital, Hong Kong
3 Department of Ophthalmology, Tseung Kwan O Hospital, Hong Kong
Corresponding author: Dr Cynthia KL Cheng (chengklc@yahoo.com)
 Full paper in PDF
Full paper in PDF
Case report
In January 2020, a 32-year-old Filipino woman was
admitted to the medical unit of our hospital with
subacute onset of a facial malar rash, digital vasculitis
purpura and alopecia. Systemic lupus erythematosus
(SLE) had been newly diagnosed according to the
Systemic Lupus International Collaborating Clinics
criteria. Initial blood tests revealed an elevated
serum anti-ds DNA antibody level, positive anti-SM
antibody, low C3 and C4 as well as pancytopenia.
Fasting glucose and lipid profile were unremarkable.
She was first treated with oral hydroxychloroquine
200 mg daily and oral prednisolone 20 mg daily.
One week later, she presented to our ophthalmology
clinic with acute-onset painless blurring of vision in
her right eye upon waking that morning. Physical
examination revealed a best-corrected visual acuity
of finger counting at 30 cm in her right eye and 6/6
in her left eye with a right relative afferent pupillary
defect. Slit-lamp examination was unremarkable
with no anterior chamber inflammation. Fundus
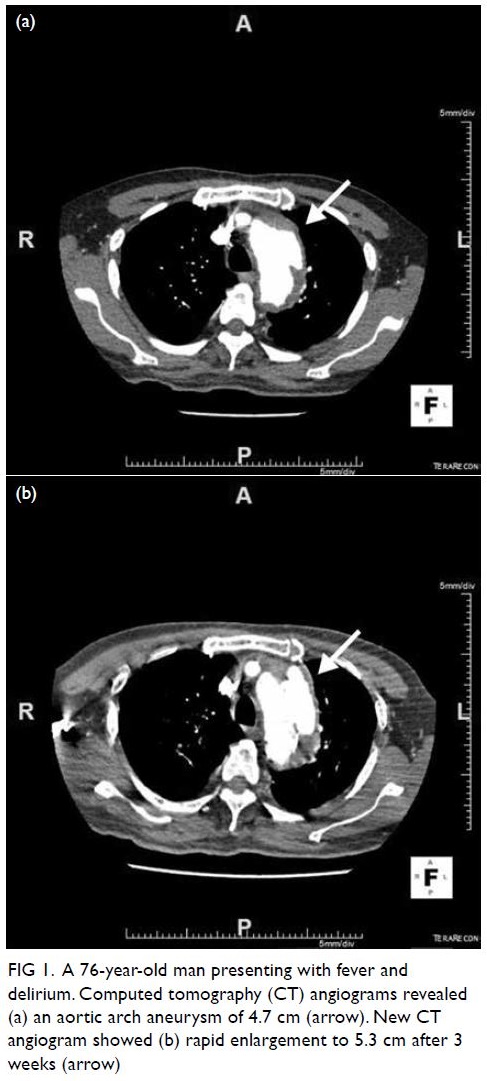
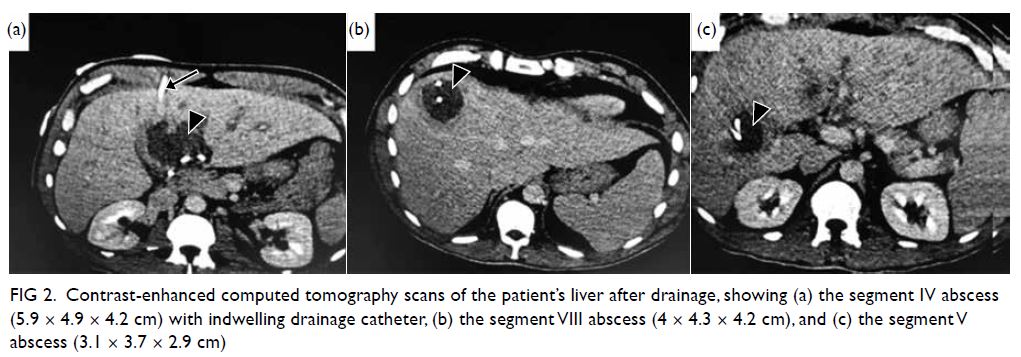
examination revealed numerous Purtscher flecken,
which are polygonal patches of retinal whitening
with distinct border and normal retina in between,
across the posterior pole. A pseudo cherry-red
spot and confluent retinal whitening could be seen
in the right eye secondary to proximal occlusion
of the retinal arteries. A few flame-shaped retinal
haemorrhages were observed inferior and temporal
to the optic disc in the right eye. The right optic disc
was mildly pale with sharp margins and the retinal
vessels were not tortuous, while the left optic disc is
pink (Fig 1).
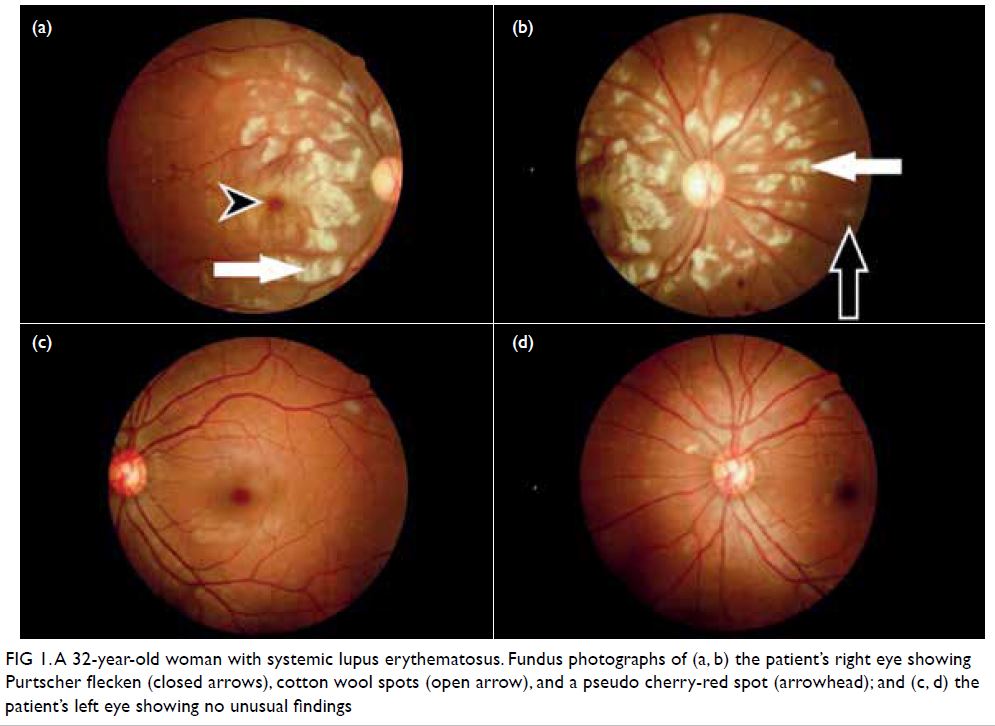
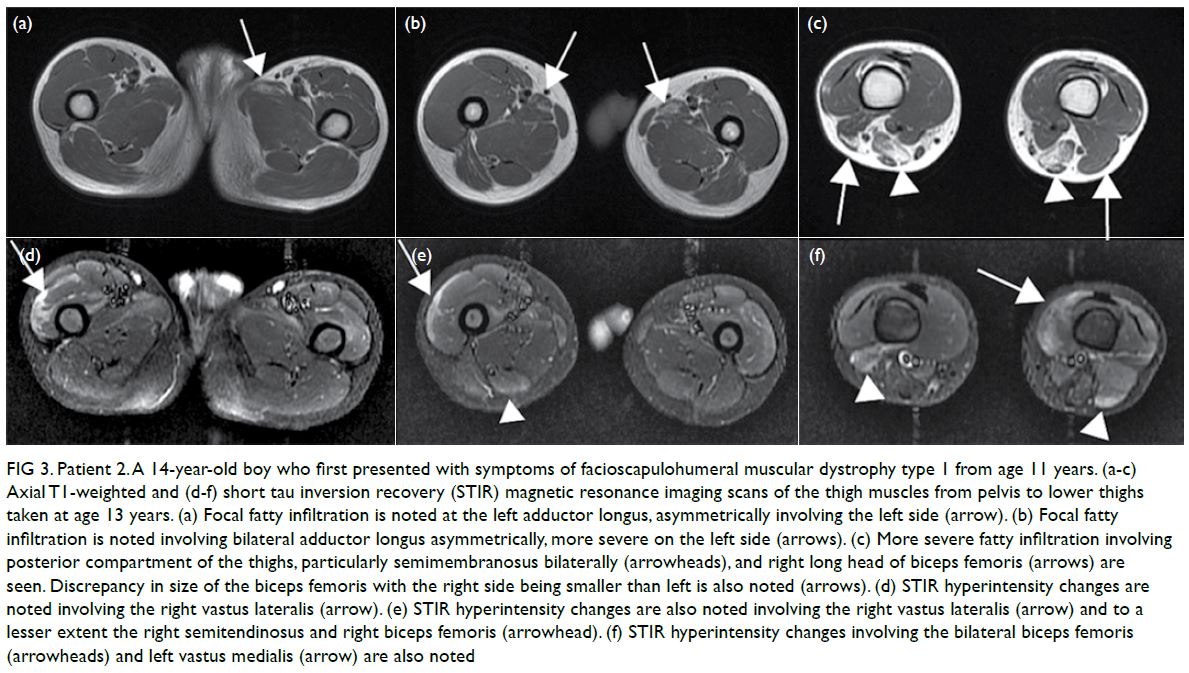
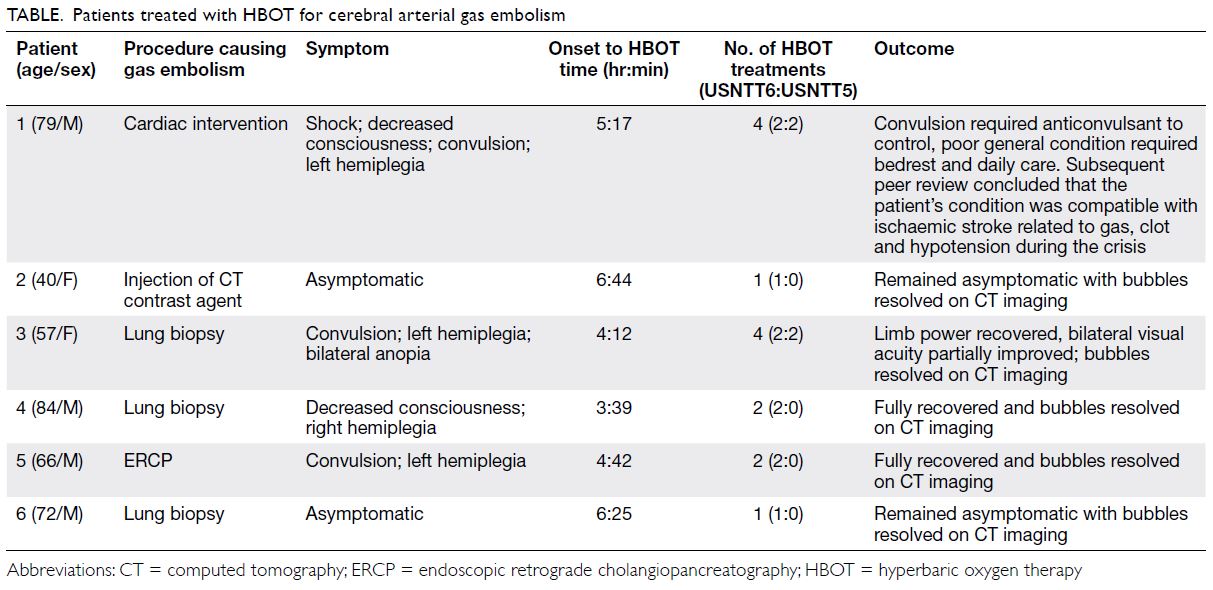
Figure 1. A 32-year-old woman with systemic lupus erythematosus. Fundus photographs of (a, b) the patient’s right eye showing Purtscher flecken (closed arrows), cotton wool spots (open arrow), and a pseudo cherry-red spot (arrowhead); and (c, d) the patient’s left eye showing no unusual findings
Urgent optical coherence tomography of the
right eye showed hyper-reflectivity in the retinal
nerve fibre layer and areas of retinal thickening.
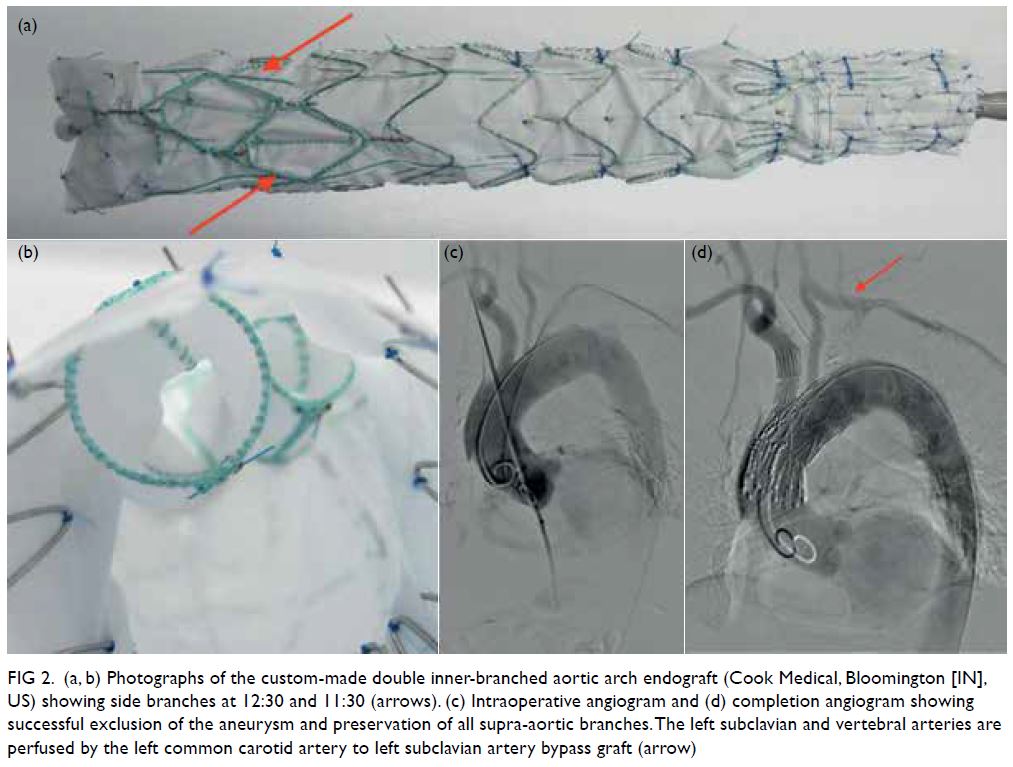
Fundus fluorescein angiography (FFA) of the
patient’s right eye showed capillary non-perfusion
corresponding to Purtscher flecken, superior branch
retinal arterial occlusion, and peripapillary staining
(Fig 2). Optical coherence tomography and FFA
of the patient’s left eye were normal. Given the typical fundus appearance and FFA findings, our
provisional diagnosis was Purtscher-like retinopathy
with branch retinal artery occlusion related to
SLE. We advised her medical team to escalate her
systemic treatment immediately and she was given
intravenous methylprednisolone 500 mg daily for
3 days, followed by oral cyclophosphamide 500 mg
for 1 day before switching to oral prednisolone
40 mg daily. Oral hydroxychloroquine 200 mg daily
was continued. On high-dose steroids, she developed
features of psychosis that subsequently resolved.
Magnetic resonance imaging of the brain showed no
features typical of central nervous system lupus such
as ischaemia and vasculitis. We closely monitored
her condition and visual acuity had not improved
1 month after systemic treatment was started,
remaining at finger counting at 30 cm. Fundus
examination of the right eye after treatment revealed
macula oedema and pseudo cherry-red spot, with no
signs of optic atrophy. Cotton wool spots in the left
eye had resolved. The patient chose to seek further
medical attention overseas.
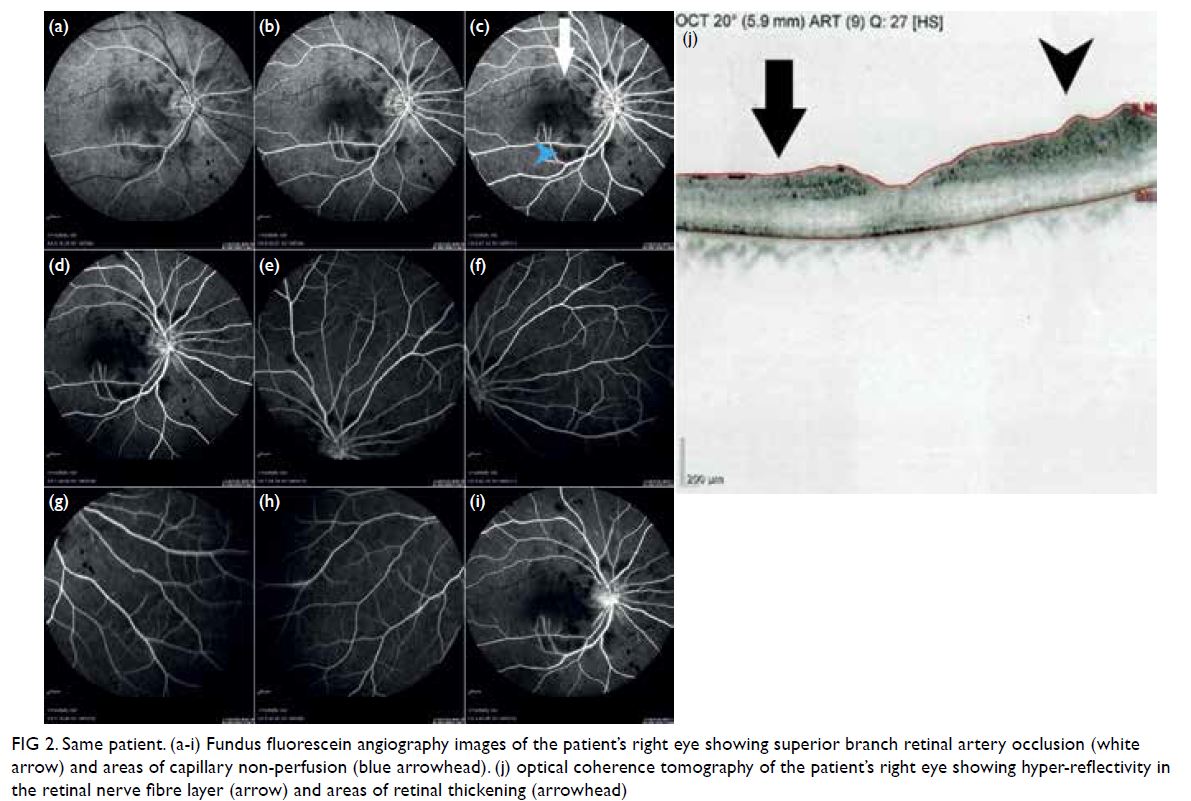
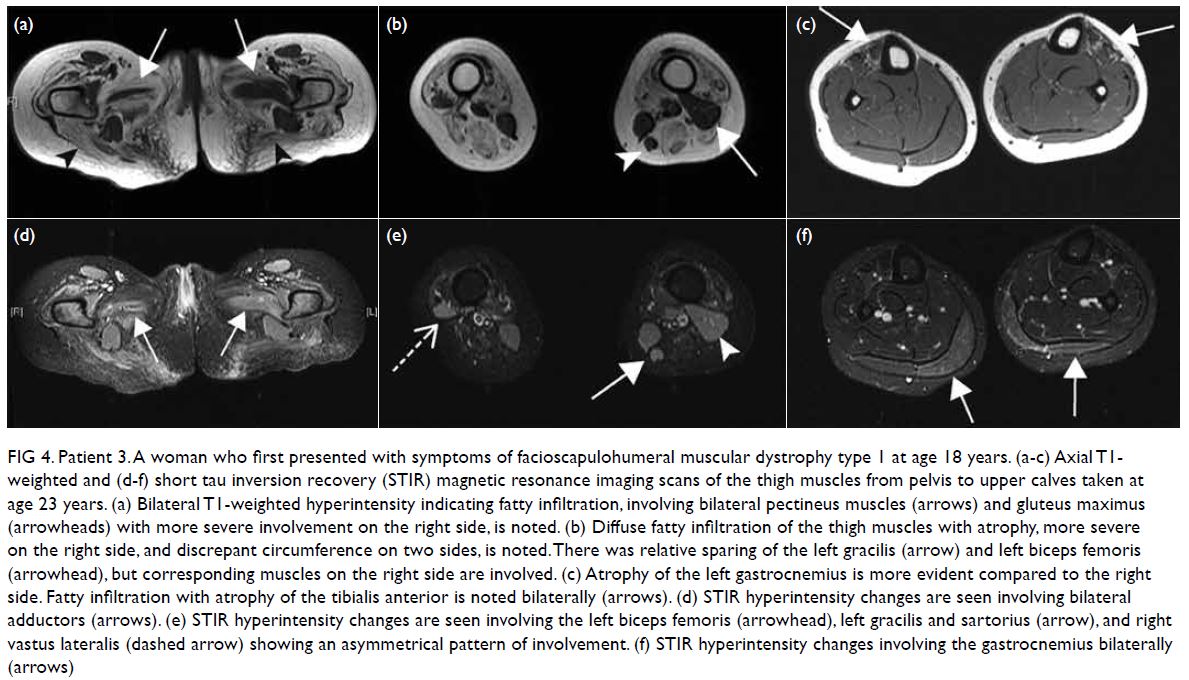
Figure 2. Same patient. (a-i) Fundus fluorescein angiography images of the patient’s right eye showing superior branch retinal artery occlusion (white arrow) and areas of capillary non-perfusion (blue arrowhead). (j) optical coherence tomography of the patient’s right eye showing hyper-reflectivity in the retinal nerve fibre layer (arrow) and areas of retinal thickening (arrowhead)
Discussion
At least one third of patients with SLE have
ophthalmological involvement. Retinal and choroidal
pathology are sinister ocular complications of SLE
that can lead to permanently impaired visual acuity.
Purtscher-like retinopathy is a type of vaso-occlusive
retinopathy associated with multiple conditions
including connective tissue disorders, pancreatic
disease, renal disease, and haematological disease.1
We report this rare case of Purtscher-like
retinopathy in a patient with SLE which had a
devastating visual outcome despite systemic
treatment. In an observational case series of
5688 patients with SLE, eight cases of Purtscher-like
retinopathy were diagnosed.1 All patients had
received treatment but most had optic atrophy
and persistent low visual acuity,1 consistent with
the outcome for our patient. Poor visual acuity has
also been attributed to presentation with a pseudo cherry-red spot. The prognosis of Purtscher-like
retinopathy is better in patients with other underlying
diseases. For example, Shahlaee et al2 reported a case
of postviral Purtscher-like retinopathy presenting
with finger count visual acuity in both eyes. No
treatment was given but the patient’s final visual
acuity improved to 20/20 with bilateral scotoma on
visual field assessment. Another case of Purtscher-like
retinopathy in a patient with adult-onset Still’s
Disease was reported by Yachoui,3 in which initial
visual acuity was 20/20 in the right eye and 20/25 in
the left. Oral prednisolone 60 mg daily was initiated
and the patient’s final visual acuity was 20/25 in both
eyes with evidence of established bilateral optic
atrophy.
In a retrospective case control study, Gao et al4
reported a 0.66% prevalence of retinal vasculopathy
among SLE patients with signs ranging from cotton
wool spots, retinal vascular attenuation to retinal
haemorrhages. Lupus retinopathy can result in
severe complications such as neovascularisation,
macula oedema and retinal vessel occlusion resulting
in vision loss.4 It is known that patients with high
Systemic Lupus Erythematosus Activity Disease
Index score are at higher risk of Purtscher-like
retinopathy, central nervous system lupus1 and vaso-occlusive retinopathy, reflecting severe systemic
microangiopathy.1 4
Because patients with active lupus are at risk
of Purtscher-like retinopathy that can lead to severe
irreversible visual loss,1 it is important to raise
awareness in patients with active lupus and visual
symptoms. A high index of suspicion for retinopathy
is needed for those with very active disease and
prompt referral to an ophthalmologist is warranted in
the presence of visual symptoms. Despite escalation
of treatment after the onset of visual symptoms,
our patient’s vision did not recover. Early aggressive
treatment to control SLE disease activity is essential
to prevent this blinding complication.
References
1. Wu C, Dai R, Dong F, Wang Q. Purtscher-like retinopathy in systemic lupus erythematous. Am J Ophthalmol
2014;158:1335-1341.e1. Crossref
2. Shahlaee A, Sridhar J, Rahimy E, Shieh WS, Ho AC. Paracentral acute middle maculopathy associated with
post viral Purtshcer-like retinopathy. Retinal Cases Brief
Rep 2019;13:50-3. Crossref
3. Yachoui R. Purtscher-like retinopathy associated with adult-onset
still disease. Retin Cases Brief Rep 2018;12:379-81. Crossref
4. Gao N, Li MT, Li YH, et al. Retinal vasculopathy in patients with systemic lupus erythematous. Lupus 2017;26:1182-9. Crossref