Posaconazole-induced gynaecomastia in a patient with COVID-19–associated mucormycosis: a case report
Hong Kong Med J 2025 Aug;31(4):316–8 | Epub 6 Aug 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Posaconazole-induced gynaecomastia in a patient with COVID-19–associated mucormycosis: a case report
Mohammadreza Salehi, MD1; Hossein Khalili, PharmDM2; Amirmasoud Kazemzadeh, MD3; Maryam Alaei, PharmD2; Azin Tabari, MD4
1 Research Centre for Antibiotic Stewardship and Antimicrobial Resistance, Department of Infectious Diseases, Imam Khomeini Hospital
Complex, Tehran University of Medical Sciences, Tehran, Iran
2 Department of Clinical Pharmacy, School of Pharmacy, Tehran University of Medical Sciences, Tehran, Iran
3 Department of Internal Medicine, School of Medicine, Imam Khomeini Hospital Complex, Tehran University of Medical Sciences, Tehran, Iran
4 Department of Ear, Nose, and Throat Diseases, School of Medicine, Imam Khomeini Hospital Complex, Tehran University of Medical Sciences, Tehran, Iran
Corresponding author: Dr Amirmasoud Kazemzadeh (amirm.kazemzadeh@gmail.com)
 Full paper in PDF
Full paper in PDF
Case presentation
A 49-year-old man with a history of poorly controlled
diabetes for over 10 years was hospitalised 6 months
earlier, on 2 April 2023, for severe COVID-19
infection. He was discharged after 3 weeks but later
developed right-sided facial pain and diplopia. He
was diagnosed with sino-orbital mucormycosis
based on the clinical symptoms and imaging
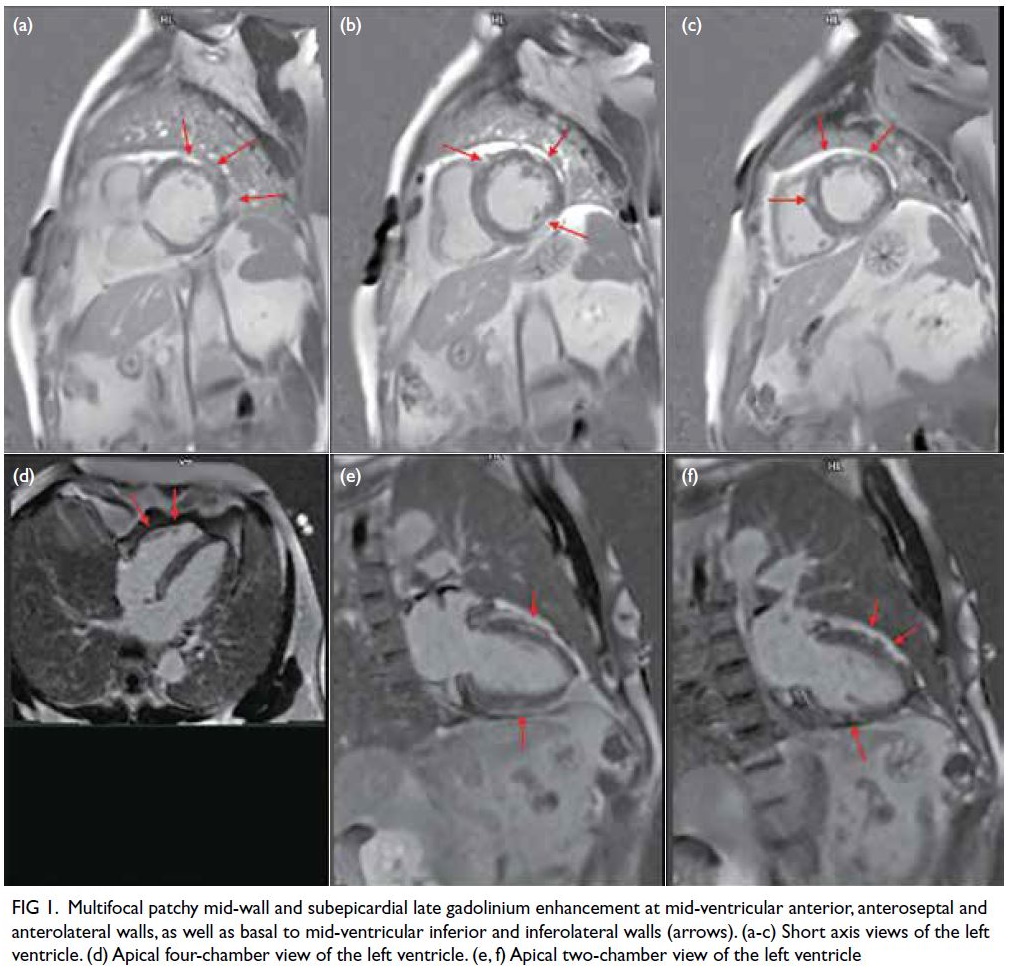
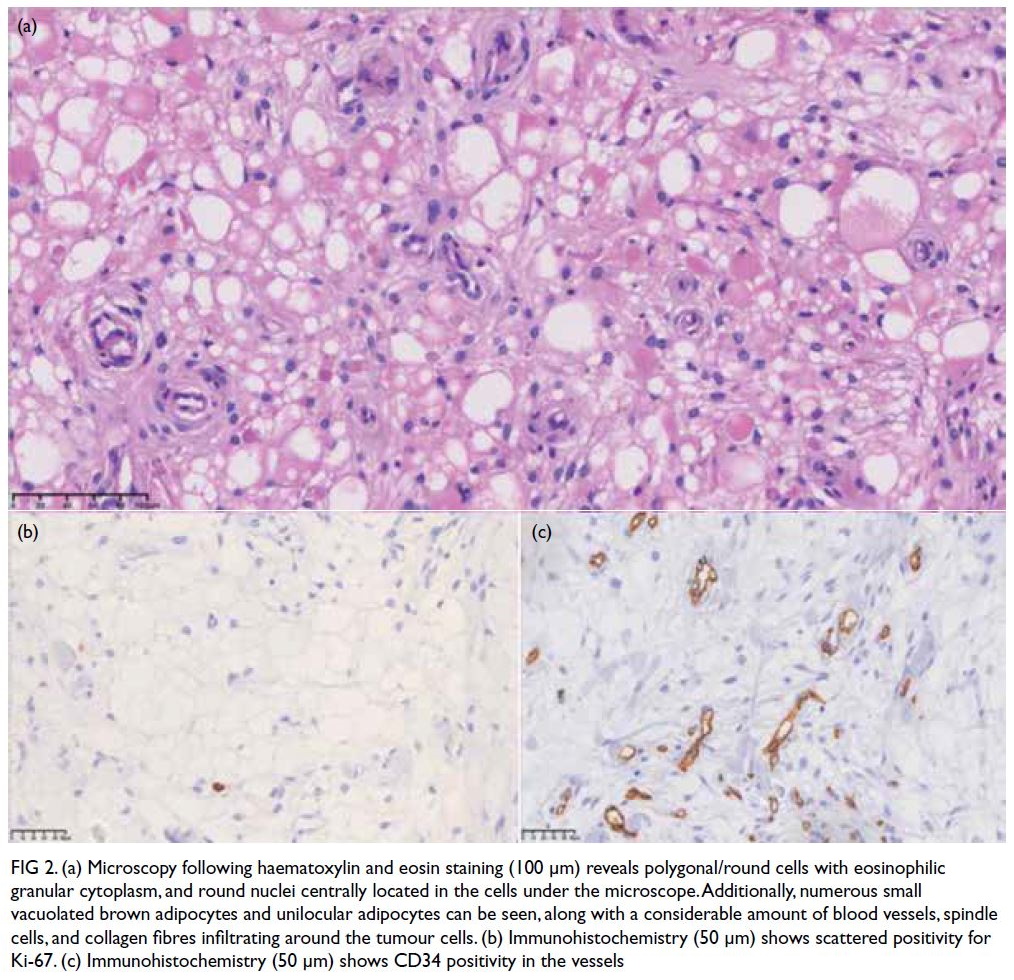
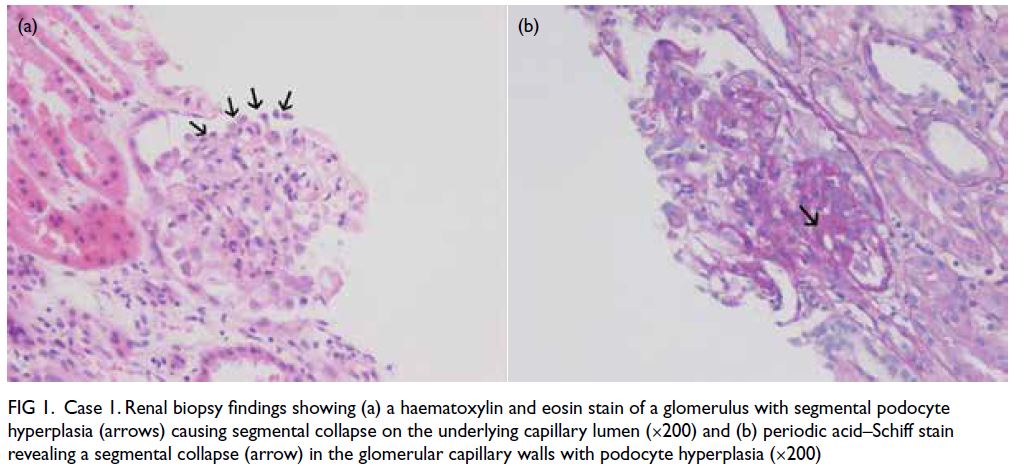
features (Fig a). Histopathological examination of a sinus biopsy revealed invasive ribbon-like hyphae,
confirming the diagnosis. The patient underwent
sinus and surgical debridement of the orbit and was
prescribed antifungal treatment. After two surgical
debridements of necrotic tissue and 6 weeks of
combination antifungal treatment with liposomal
amphotericin B (400 mg daily via infusion) plus
posaconazole (300 mg twice daily on the first day
and 300 mg daily intravenously thereafter), the
patient was discharged in good general health but
with right eye blindness. He was advised to continue
treatment with oral posaconazole (300 mg twice
daily on the first day, then 300 mg daily thereafter)
for a duration of 12 weeks and to control his blood
sugar with insulin.
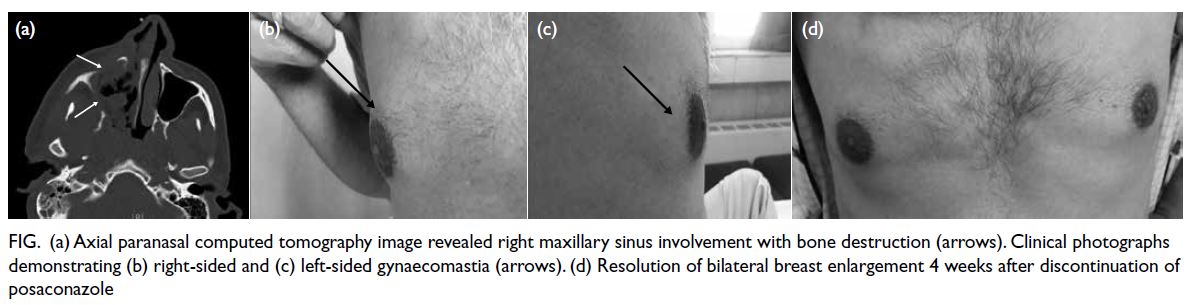
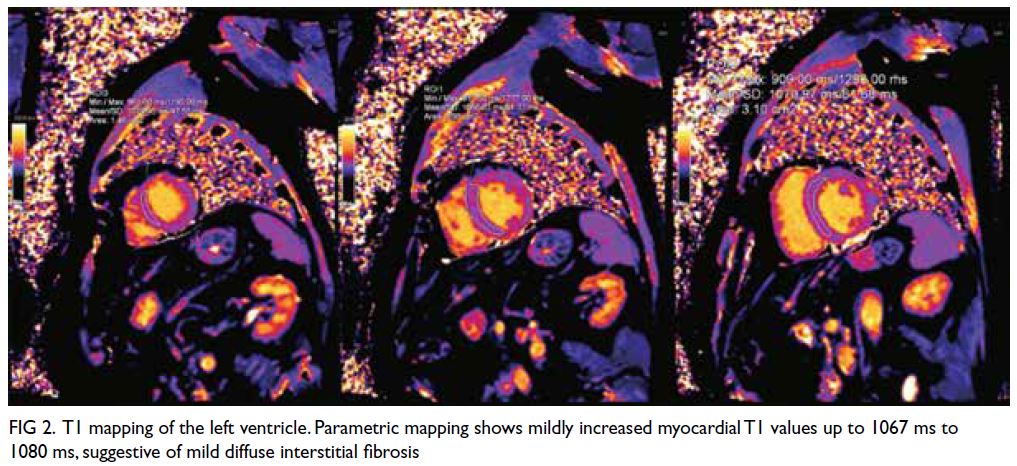
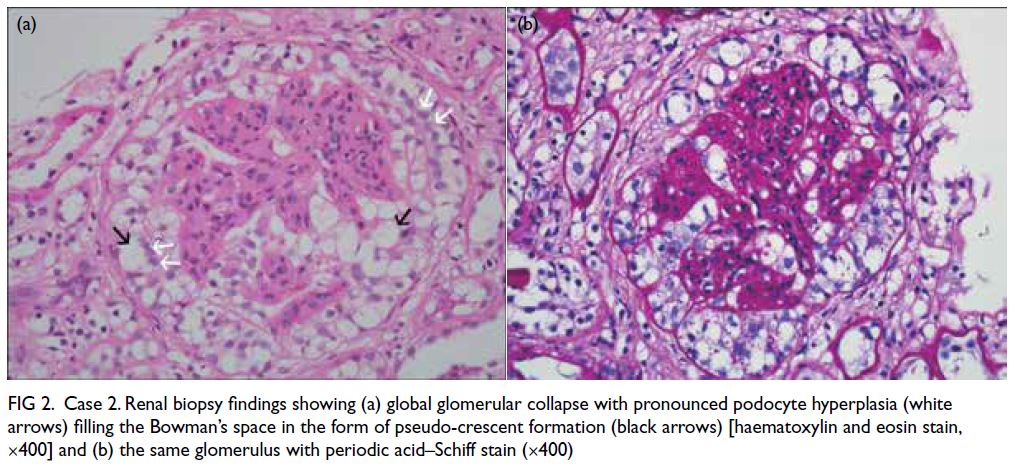
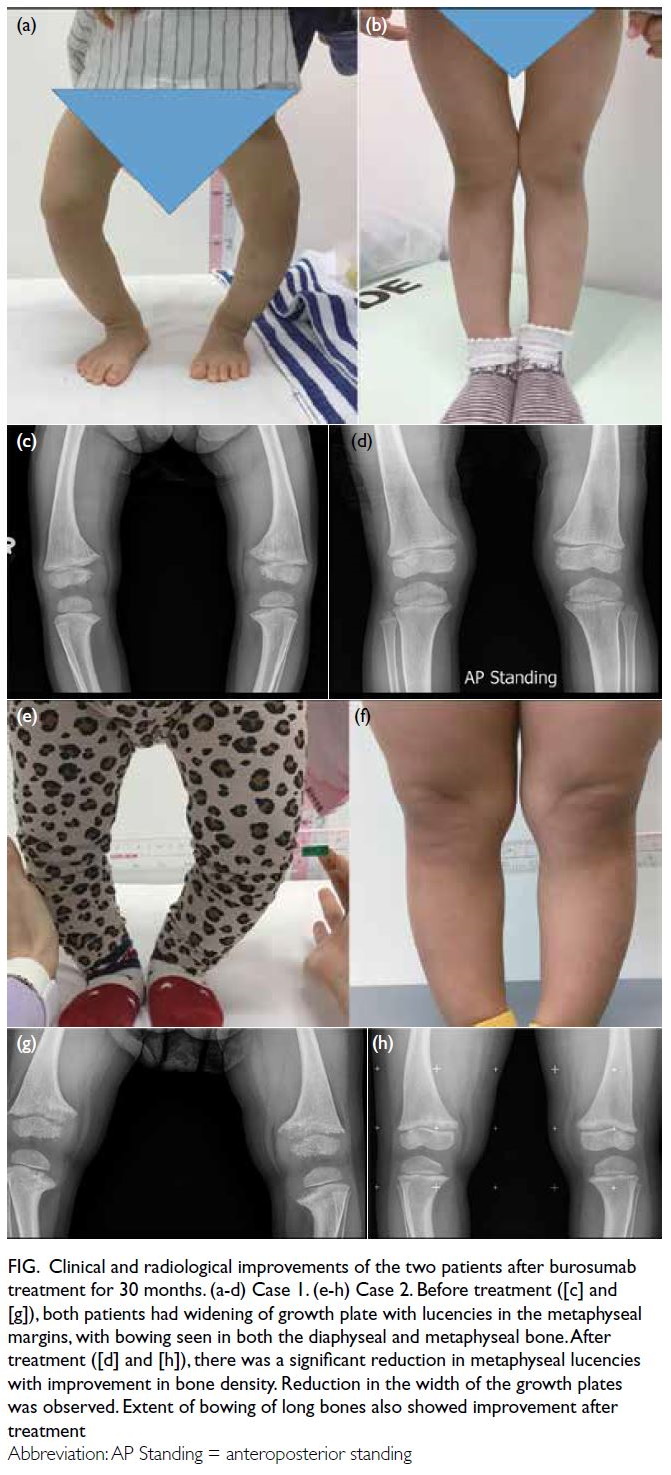
Figure. (a) Axial paranasal computed tomography image revealed right maxillary sinus involvement with bone destruction (arrows). Clinical photographs demonstrating (b) right-sided and (c) left-sided gynaecomastia (arrows). (d) Resolution of bilateral breast enlargement 4 weeks after discontinuation of posaconazole
In September 2023, 2 months after discharge,
the patient presented to the hospital for follow-up,
complaining of increased bilateral breast volume,
particularly in the right breast (Fig b and c). He had
first noticed this increase in volume approximately
1 month after hospitalisation, and it had gradually
intensified.
Clinical examination revealed bilateral breast
swelling with increased breast adipose tissue volume,
more pronounced on the right side. No tenderness
or palpable mass was detected. Ultrasound findings
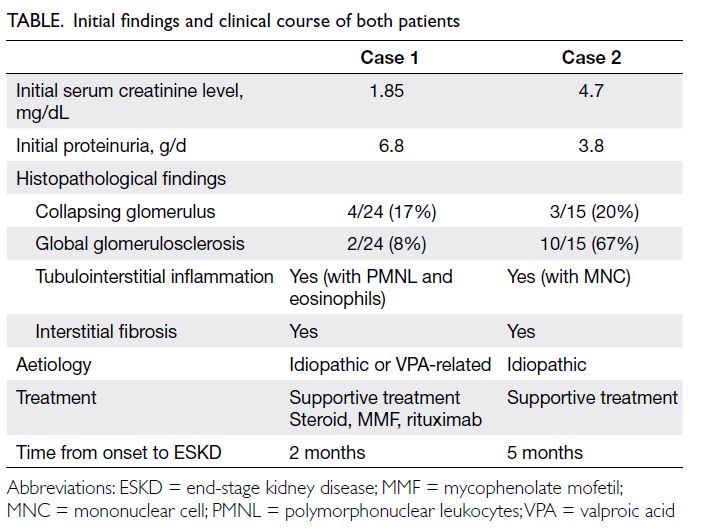
were consistent with gynaecomastia. Hormone
testing showed a normal prolactin level and pituitary
axes but markedly decreased testosterone and
elevated oestrogen levels (Table).
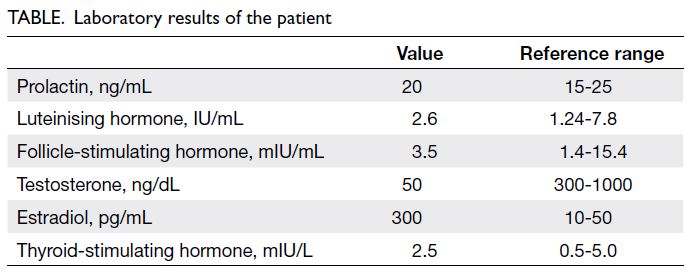
Table. Laboratory results of the patient
In view of these results and the possible
association of gynaecomastia with azole use,
posaconazole was discontinued. The patient was
prescribed liposomal amphotericin B 400 mg
daily and monitored daily to ensure compliance
with medication and to evaluate any possible side-effects.
He exhibited minor hypokalaemia while
receiving amphotericin B, which resolved with the
administration of potassium supplements. There was no evidence of hyperaldosteronism during treatment
with posaconazole. Following the discontinuation of
posaconazole, the patient’s gynaecomastia improved
significantly over 4 weeks (Fig d).
Discussion
At the time of writing, this report represents the
second documented case of posaconazole-induced
gynaecomastia. Information on this adverse effect is
limited, despite its recognised association with azole
use. Given the common use of posaconazole for the
prevention and treatment of mucormycosis, other
patients may be at risk of similar complications.
Disseminating this information is essential for
clinical pharmacists to manage such cases effectively
Our case was a middle-aged man with a
history of diabetes and COVID-19, who developed
gynaecomastia following the prescription of
posaconazole for mucormycosis. Gynaecomastia
is a benign enlargement of the breast tissue that
may occur in many adult men.1 Physiological
gynaecomastia in infants, adolescents, and older
adult men is usually unilateral. Non-physiological
gynaecomastia may arise in patients with chronic
diseases such as hypogonadism, cirrhosis,
neoplasms or uraemia, or as a consequence of drug
use. Additionally, non-physiological gynaecomastia
may cause localised pain.2 In our patient, bilateral
and painless non-physiological gynaecomastia
developed as a consequence of azole use.
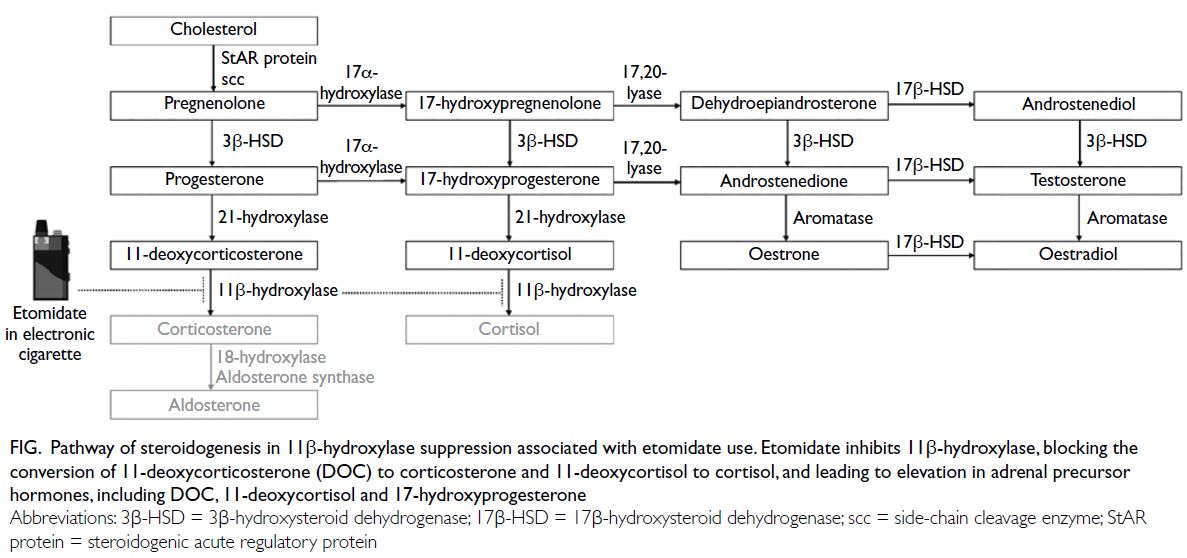
Azole antifungals are the mainstay of
prevention and treatment for invasive fungal
infections such as mucormycosis.3 Most reported
cases of azole-induced gynaecomastia have involved
patients taking ketoconazole and, to a lesser
extent, itraconazole. Both inhibit cytochrome P450
enzymes involved in steroidogenesis.4 In-vitro
evidence suggests that posaconazole can inhibit
cytochrome P450 17A1, an enzyme involved in the
lyase reaction required for androgen production. In
addition, posaconazole, similar to ketoconazole, may
inhibit hepatic oestrogen metabolism.5 Accordingly,
our patient’s serum testosterone level was lower than normal, while the oestradiol level was significantly
elevated. This is comparable to the first reported
case of posaconazole-induced gynaecomastia by
Thompson et al.6 In that patient, who was prescribed
long-term posaconazole for coccidioidomycosis
(300 mg/day slow-release formulation), the oestradiol
level was raised despite a normal testosterone level.
Notably, gynaecomastia did not improve following
a switch from posaconazole to voriconazole. In
contrast, our patient showed improvement after the
complete discontinuation of azole agents.
The main limitation of this report is the
absence of a serum posaconazole level, as testing
was unavailable due to economic constraints
affecting resource availability. This limits the ability
to draw a definitive conclusion regarding the
association of posaconazole with the development
of gynaecomastia, or the observed improvement
following its discontinuation.
Author contributions
Concept or design: M Salehi, H Khalili.
Acquisition of data: A Kazemzadeh, M Alaei.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: M Salehi, A Tabari.
Acquisition of data: A Kazemzadeh, M Alaei.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: M Salehi, A Tabari.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Acknowledgement
The authors thank Mrs Fariba Zamani for language editing of the manuscript.
Funding/support
This study received no specific grant from any funding agency
in the public, commercial, or not-for-profit sectors.
Ethics approval
This study was approved of by the institutional review
board of Tehran University of Medical Sciences, Iran (Ref
No.: IR.TUMS.IKHC.REC.1403.165). Written consent was
obtained from the patient for publication of this case report.
References
1. Fagerlund A, Lewin R, Rufolo G, Elander A, Santanelli di
Pompeo F, Selvaggi G. Gynecomastia: a systematic review.
J Plast Surg Hand Surg 2015;49:311-8. Crossref
2. Jin Y, Fan M. Treatment of gynecomastia with prednisone:
case report and literature review. J Int Med Res
2019;47:2288-95. Crossref
3. Benitez LL, Carver PL. Adverse effects associated with
long-term administration of azole antifungal agents. Drugs
2019;79:833-53. Crossref
4. Satoh T, Fujita KI, Munakata H, et al. Studies on the
interactions between drugs and estrogen: analytical method for prediction system of gynecomastia induced
by drugs on the inhibitory metabolism of estradiol using
Escherichia coli coexpressing human CYP3A4 with human
NADPH-cytochrome P450 reductase. Anal Biochem
2000;286:179-86. Crossref
5. Yates CM, Garvey EP, Shaver SR, Schotzinger RJ, Hoekstra WJ. Design and optimization of highly-selective, broad
spectrum fungal CYP51 inhibitors. Bioorg Med Chem Lett
2017;27:3243-8. Crossref
6. Thompson GR 3rd, Surampudi PN, Odermatt A.
Gynecomastia and hypertension in a patient treated with
posaconazole. Clin Case Rep 2020;8:3158-61. Crossref