Use of burosumab in two young children with X-linked hypophosphataemic rickets in Hong Kong: two case reports
Hong Kong Med J 2024 Dec;30(6):506–8 | Epub 26 Nov 2024
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Use of burosumab in two young children with X-linked hypophosphataemic rickets in Hong Kong: two case reports
Joanna YL Tung, MB, BS, FHKAM (Paediatrics)1; Sammy Wong, MB, ChB, FHKAM (Paediatrics)2; Queenie WS See, MB, BS, FHKAM (Paediatrics)3; Joyce Chan, MB, ChB, FHKAM (Radiology)4; Evelyn Kuong, MB, BS, FHKAM (Orthopaedics)5
1 Department of Paediatrics and Adolescent Medicine, Hong Kong Children’s Hospital, Hong Kong SAR, China
2 Department of Paediatrics and Adolescent Medicine, Alice Ho Miu Ling Nethersole Hospital, Hong Kong SAR, China
3 Department of Paediatrics and Adolescent Medicine, Queen Mary Hospital, School of Clinical Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, China
4 Department of Radiology, Hong Kong Children’s Hospital, Hong Kong SAR, China
5 Department of Orthopaedics and Traumatology, Hong Kong Children’s Hospital and Duchess of Kent Children’s Hospital, Hong Kong SAR, China
Corresponding author: Dr Joanna YL Tung (tyl404@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Case presentations
Two Chinese girls (22 months and 26 months of
age [Case 1 and Case 2, respectively]) presented in
August 2021 and November 2021 with progressive
bowing of the lower limbs since infancy. Neither had
any significant family history. Physical examination
revealed rachitic deformity, waddling gait, and
short stature. Further investigations showed
hypophosphataemia, isolated hyperphosphaturia
and a high serum alkaline phosphatase (ALP) level
with normal serum calcium, 25(OH) vitamin D,
and parathyroid hormone levels. Characteristic
clinical and radiographical findings suggestive
of hypophosphataemic rickets were also evident
(Fig). Molecular testing confirmed a diagnosis
of X-linked hypophosphataemia (XLH) with
heterozygous pathogenic PHEX (phosphate-regulating
endopeptidase homologue on the X
chromosome) variants detected in both patients
[Case 1: c.2104C>T, p.(Arg702); Case 2: (c.1699c>T)
(Arg567)]. Neither set of parents was affected. Both
girls were commenced on conventional treatment
with phosphate solution and alfacalcidol for around
3 months with only fair improvement. They were
then started on burosumab at a starting dose of
0.8 mg/kg every 2 weeks, titrated up to 2 mg/kg
(20 mg) 4 months later based on fasting serum
phosphate level. Despite maximum doses of
burosumab, serum phosphate level remained
slightly below the normal range for both patients.
Nevertheless there was ongoing clinical improvement
in ALP level, the ratio of tubular maximum
reabsorption of phosphate to glomerular filtration
rate, growth velocity, and healing of rickets in both
patients. After burosumab treatment for 24 months,
both patients had significant clinical, biochemical,
and radiological improvement (Table and Fig). They were followed up by the multidisciplinary team at
the Hong Kong Children’s Hospital. No treatment-related
adverse reactions or disease-related
complications (including skull deformities, dental
abscess, and hearing problem) were observed.
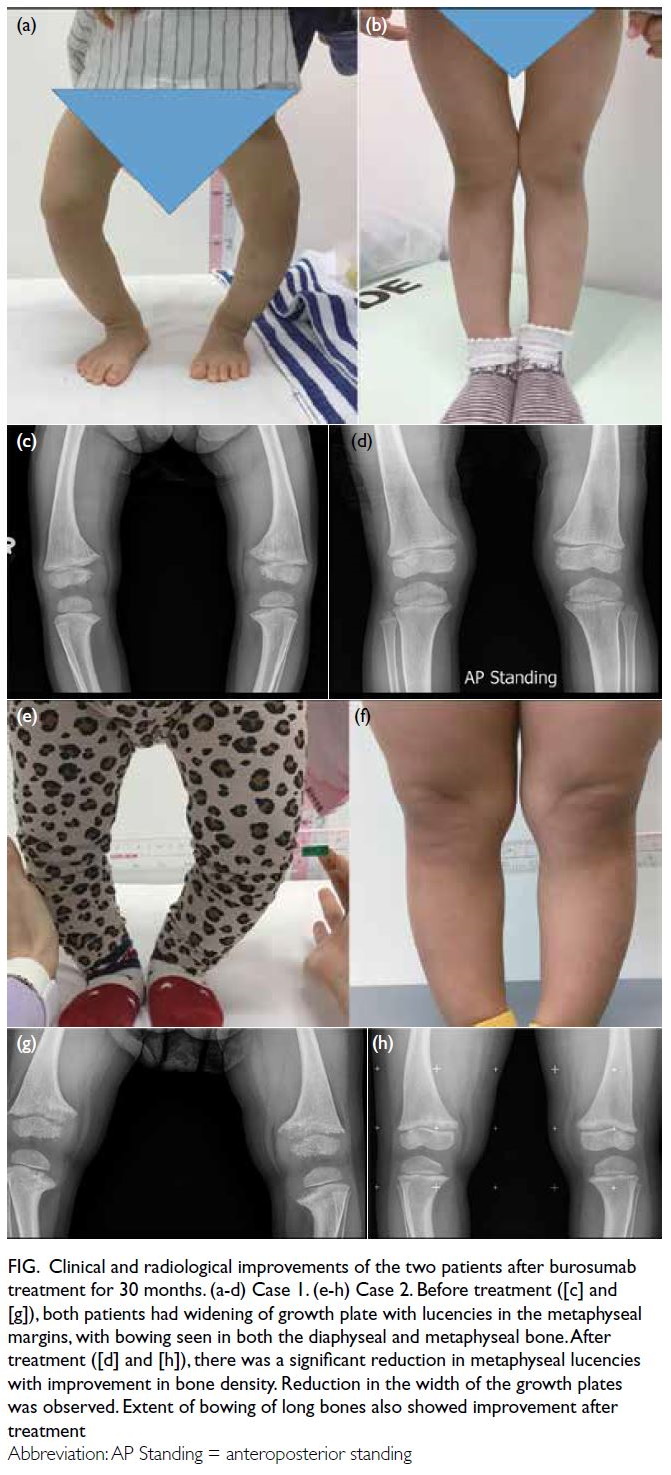
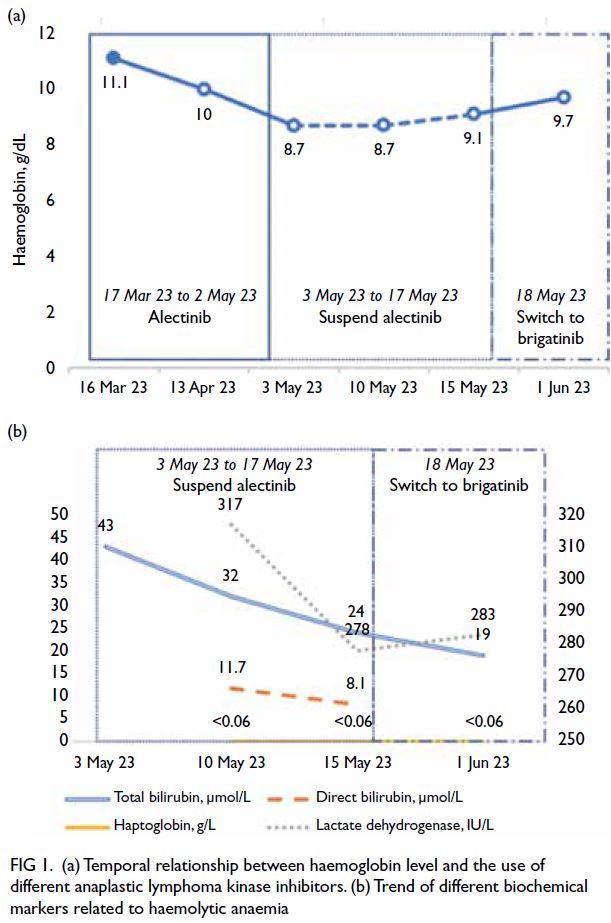
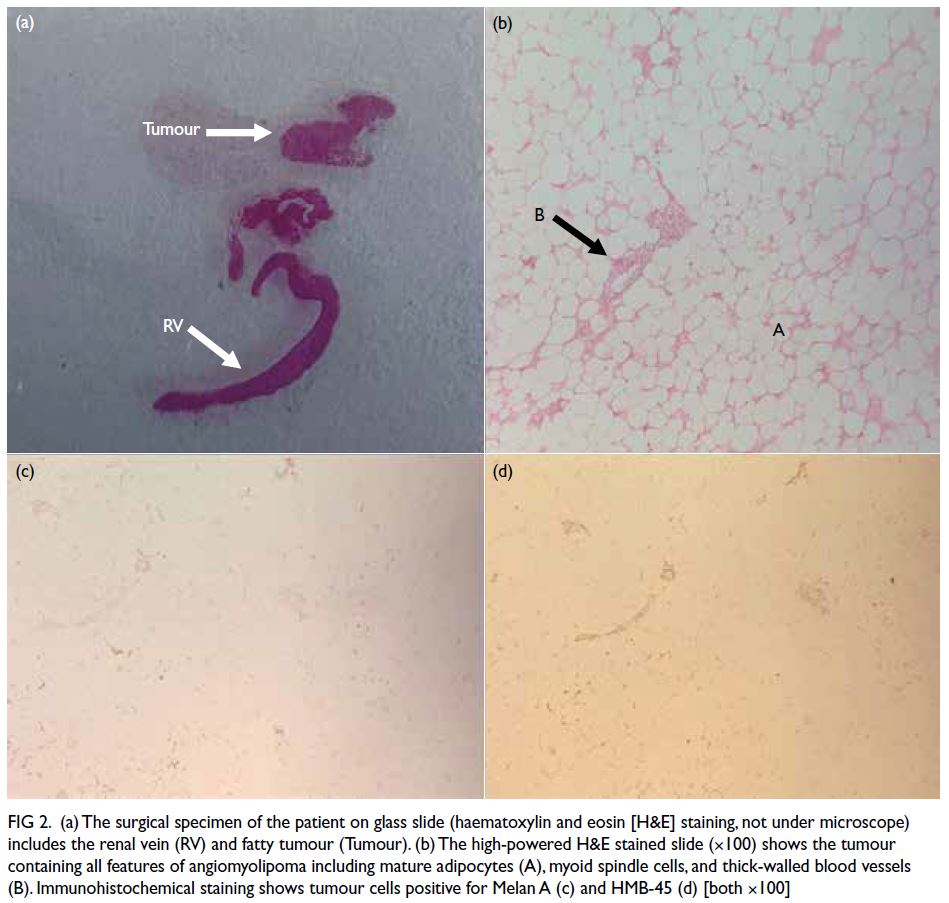
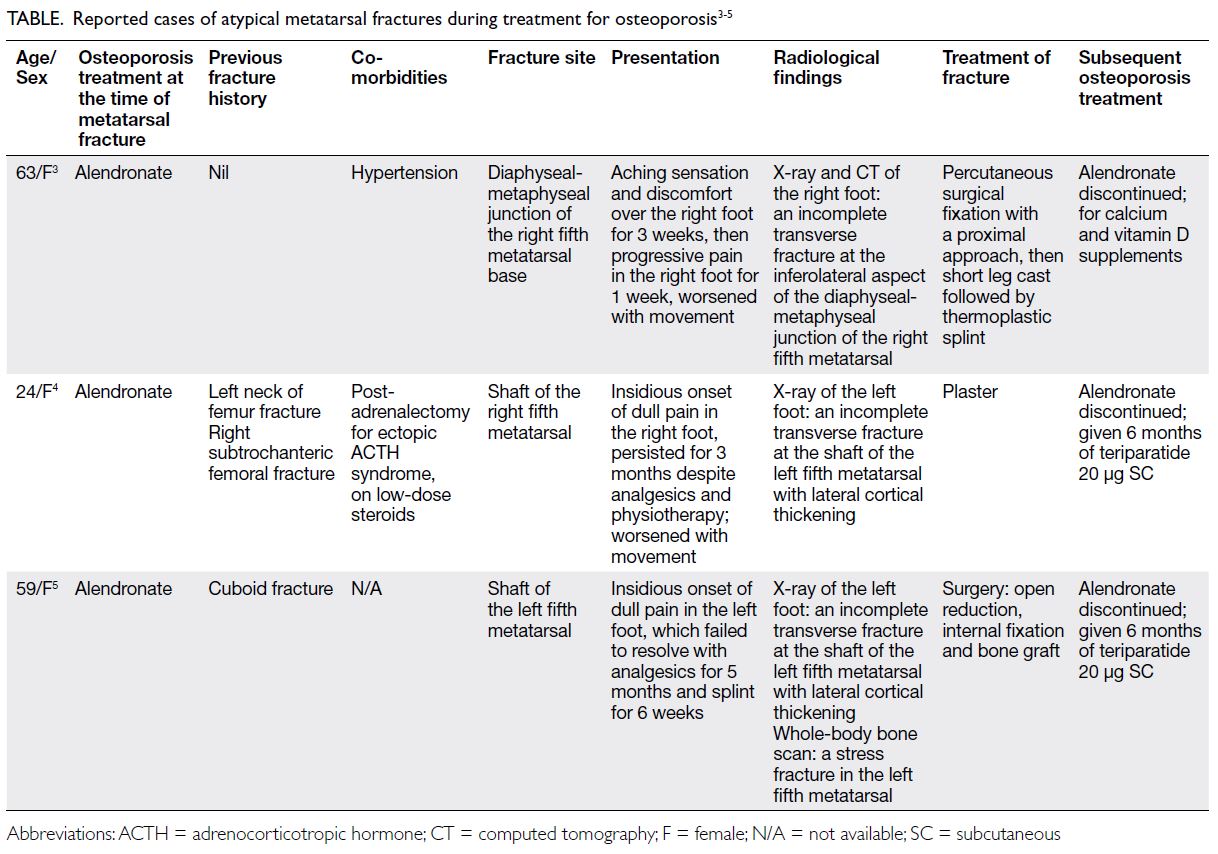
Figure. Clinical and radiological improvements of the two patients after burosumab treatment for 30 months. (a-d) Case 1. (e-h) Case 2. Before treatment ([c] and [g]), both patients had widening of growth plate with lucencies in the metaphyseal margins, with bowing seen in both the diaphyseal and metaphyseal bone. After treatment ([d] and [h]), there was a significant reduction in metaphyseal lucencies with improvement in bone density. Reduction in the width of the growth plates was observed. Extent of bowing of long bones also showed improvement after treatment.
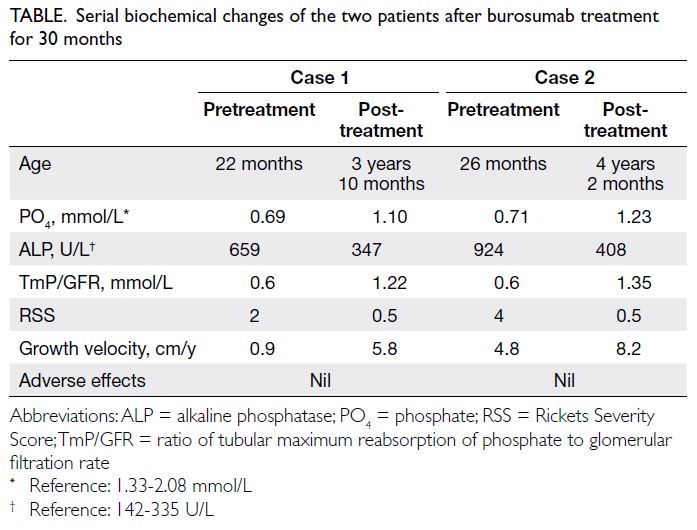
Table. Serial biochemical changes of the two patients after burosumab treatment for 30 months
Discussion
X-linked hypophosphataemia is the most common
cause of genetic rickets, with a prevalence of 1:20 000
to 1:60 000.1 2 It is caused by mutations in the
PHEX gene and results in an increased level of the
fibroblast growth factor 23 (FGF-23) hormone. This
leads to impaired renal reabsorption of phosphate,
low serum 1,25-dihydroxyvitamin D concentration,
and reduced intestinal phosphate absorption, and
ultimately, chronic hypophosphataemia. Patients
typically present in early childhood with signs and
symptoms of rickets and osteomalacia, progressive
bowing deformities of the lower limbs, bone pain
and stunted growth, as in our patients.
X-linked hypophosphataemia is conventionally
treated with oral phosphate and active vitamin D
analogues but this does not address the underlying
pathogenesis and may only partially correct the
biochemical derangement and skeletal deformities.
Patients often require repeated orthopaedic
procedures, eg, hemiepiphysiodesis or even
multiple corrective osteotomies to maintain the
mechanical axis of the lower limbs. Surgical
correction is fraught with technical difficulties due to
osteomalacia and there are high rates of recurrence.
Moreover, conventional treatment is associated with
long-term side-effects such as hyperparathyroidism
and nephrocalcinosis.
Burosumab, a monoclonal antibody to FGF-23
for XLH treatment, was approved by the United
States Food and Drug Administration3 and the European Medicines Agency4 in 2018. Subsequently,
a phase III trial to evaluate 61 children with XLH
aged 1 to 12 years for 64 weeks5 and another trial
evaluating children with XLH aged 5 to 12 years
for 160 weeks6 further supported its safety and
effectiveness in terms of improved total Rickets
Severity Score and fasting serum phosphate level.
These outcomes were also measured in our patients
and similar improvements observed. With the
approval of burosumab, there has been a paradigm
shift in the treatment of XLH in Western countries.
Nevertheless conventional treatment continues to
be the norm in most parts of our region—partly
attributed to the high cost of burosumab, and partly
due to the lack of published regional experience with
burosumab in the clinical setting.
In our patients, the dose of burosumab was
titrated up slowly to the maximum dose based on
age-specific fasting phosphate level, as per various
clinical practice recommendations.7 8 With the
maximum dose, fasting serum phosphate level
improved but failed to reach that of the age-specific
normal range. Other secondary causes including
vitamin D deficiency and hyperparathyroidism
were excluded. Nevertheless improvement in other
clinical parameters including serum ALP level, the
ratio of tubular maximum reabsorption of phosphate
to glomerular filtration rate, growth velocity, and
clinical and radiological evidence of rickets healing
were observed. We have maintained our two
patients on the same dose with ongoing clinical
improvements observed. This is consistent with the
recently published opinion based on early experience
in seven European countries, recommending that a
serum phosphate concentration below the age- and
sex-specific range may be acceptable and the same
maintenance dose continued as improvements in
other clinical parameters are maintained.9 This is
based largely on expert opinion with no data on
the proportion of patients who do not achieve a
normalised phosphate level and no comparison of
clinical outcomes.
X-linked hypophosphataemia is a rare,
genetic multisystem disease. In addition to the
skeletal manifestations, around two-thirds of
affected individuals have associated dental and
periodontal issues, such as spontaneous periapical
abscesses, related to poor dentin mineralisation.10
Some patients also have other complications
including craniosynostosis and impaired hearing.
Consequently, most guidelines recommend a
multidisciplinary approach for this group of
patients.7 8 Despite treatment with burosumab, some
complications cannot be mitigated. Dental abscess,
a well-known complication in patients with XLH, has not been consistently shown to be ameliorated
by burosumab treatment. In the phase 3 burosumab
trial,5 dental abscess was observed at a higher rate in
the treatment group than the control arm. This implies
that the pathophysiology of dental abscess is not
mediated only by the FGF-23 pathway. This highlights
the importance of a multidisciplinary approach
to the management of XLH, as recommended by
recent international clinical practice guidelines7 and
regional consensus statements.8 At the Hong Kong
Children’s Hospital, this group of patients is seen in
the multidisciplinary bone clinic with input from a
paediatric endocrinologist, orthopaedic surgeon,
geneticist, dental surgeon, radiologist, and case
manager (nurse practitioner). This orchestrated,
coordinated multidisciplinary care is particularly
important to maximise the effect and overall clinical
outcome of burosumab treatment that remains
remarkably expensive.
To the best of our knowledge, this is the first
report of real-world experience of XLH treated with
burosumab outside clinical trials in Asia. The target
of a normal phosphate level may not be achievable
in practice, and treatment response should also be
guided by other clinical parameters. Further research
into factors that affect the biochemical outcome
and the clinical response of groups with different
biochemical outcome is needed. A multidisciplinary
approach should be adopted in the care of children
with XLH.
Author contributions
Concept or design: JYL Tung.
Acquisition of data: JYL Tung.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: JYL Tung.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: JYL Tung.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: JYL Tung.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Declaration
Preliminary results for these two cases were presented as a poster at the 12th Asia Pacific Paediatric Endocrine Society Biennial Scientific Meeting 2022 (South Korea, 5-8 October
2022).
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patients were treated in accordance with the Declaration of Helsinki. Consent for all treatments, procedures, and consent for publication was obtained from parents of the
patients.
References
1. Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K,
Jensen TK. Incidence and prevalence of nutritional and
hereditary rickets in southern Denmark. Eur J Endocrinol
2009;160:491-7. Crossref
2. Rafaelsen S, Johansson S, Ræder H, Bjerknes R. Hereditary
hypophosphatemia in Norway: a retrospective population-based
study of genotypes, phenotypes, and treatment
complications. Eur J Endocrinol 2016;174:125-36. Crossref
3. United States Food and Drug Administration. FDA
approves first therapy for rare inherited form of rickets,
x-linked hypophosphatemia. 2018 Apr 17. Available from:
https://www.fda.gov/news-events/press-announcements/fda-approves-first-therapy-rare-inherited-form-rickets-x-linked-hypophosphatemia. Accessed 28 Oct 2024.
4. European Medicines Agency. EU/3/14/1351—orphan
designation for treatment of X-linked hypophosphataemia.
Available from: https://www.ema.europa.eu/en/medicines/human/orphan-designations/eu-3-14-1351. Accessed 28 Oct 2024.
5. Imel EA, Glorieux FH, Whyte MP, et al. Burosumab
versus conventional therapy in children with X-linked
hypophosphataemia: a randomised, active-controlled,
open-label, phase 3 trial. Lancet 2019;393:2416-27. Crossref
6. Linglart A, Imel EA, Whyte MP, et al. Sustained efficacy
and safety of burosumab, a monoclonal antibody to FGF23,
in children with X-linked hypophosphatemia. J Clin
Endocrinol Metab 2022;107:813-24. Crossref
7. Sandy JL, Simm PJ, Biggin A, et al. Clinical practice
guidelines for paediatric X-linked hypophosphataemia in
the era of burosumab. J Paediatr Child Health 2022;58:762-8. Crossref
8. Munns CF, Yoo HW, Jalaludin MY, et al. Asia-Pacific consensus recommendations on X-linked
hypophosphatemia: diagnosis, multidisciplinary
management, and transition from pediatric to adult care. JBMR Plus 2023;7:e10744. Crossref
9. Mughal MZ, Baroncelli GI, de Lucas-Collantes C, et al.
Burosumab for X-linked hypophosphatemia in children
and adolescents: opinion based on early experience in
seven European countries. Front Endocrinol (Lausanne)2023;13:1034580. Crossref
10. Baroncelli GI, Mora S. X-linked hypophosphatemic
rickets: multisystemic disorder in children requiring
multidisciplinary management. Front Endocrinol
(Lausanne) 2021;12:688309. Crossref