Megacalycosis: a rare radiological finding
Hong
Kong Med J 2020 Dec;26(6):539.e1–2
Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
Megacalycosis: a rare radiological finding
CL Cho, FRCS Ed (Urol), FHKAM (Surgery)1,2; CK Shiu, FRCR, FHKAM (Radiology)3
1 Department of Surgery, Union Hospital, Hong Kong
2 SH Ho Urology Centre, Department of Surgery, The Chinese University of Hong Kong, Hong Kong
3 Medical Imaging Department, Union Hospital, Hong Kong
Corresponding author: Dr CL Cho (chochaklam@yahoo.com.hk)
 Full
paper in PDF
Full
paper in PDF
In April 2019, a 22-year-old woman was admitted to
Union Hospital, Hong Kong, with right loin pain and
fever. She had developed acute cystitis symptoms
1 week prior to the admission and was prescribed a
course of oral antibiotic. She had good past health with
no history of urinary tract infection. On admission
she had a temperature of 39°C. The abdomen was
soft and non-tender. Laboratory tests revealed a
normal white cell count of 6.09 × 109/L and normal
serum creatinine level of 54 μmol/L. The C-reactive protein level
was elevated at 45.5 mg/L. Urine tests revealed a
slightly turbid urine with elevated white blood cell count of
260 cells/µL. There was no significant growth
on urine culture. An urgent contrast computed
tomography scan revealed features suggestive of
pyelonephritis at the lower pole of the right kidney
(Fig 1). There was no obstructive urinary stone. In
addition, the imaging demonstrated an atypical right
pelvicalyceal system (Fig 2). A course of antibiotic
was prescribed with good clinical response. Repeat
urine microscopy was unremarkable 8 weeks after the initial
presentation. Follow-up computed tomography scan
confirmed resolution of right pyelonephritis but the
unusual finding of the right pelvicalyceal system
remained unchanged (Fig 3).
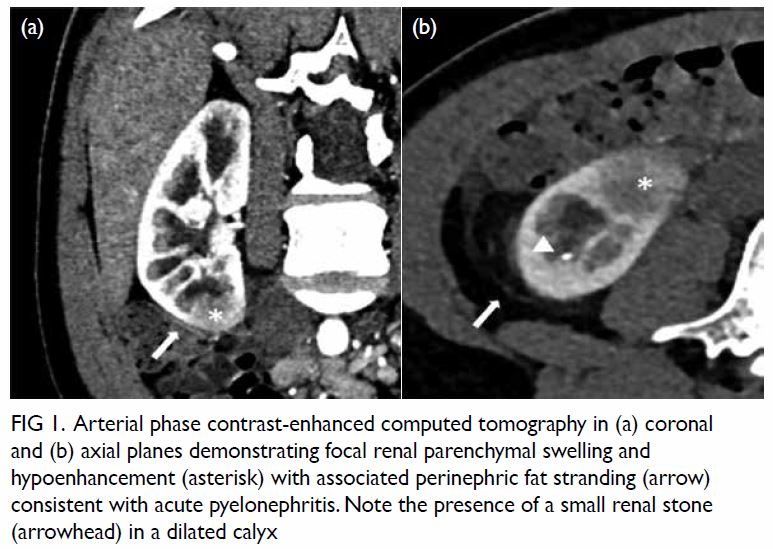
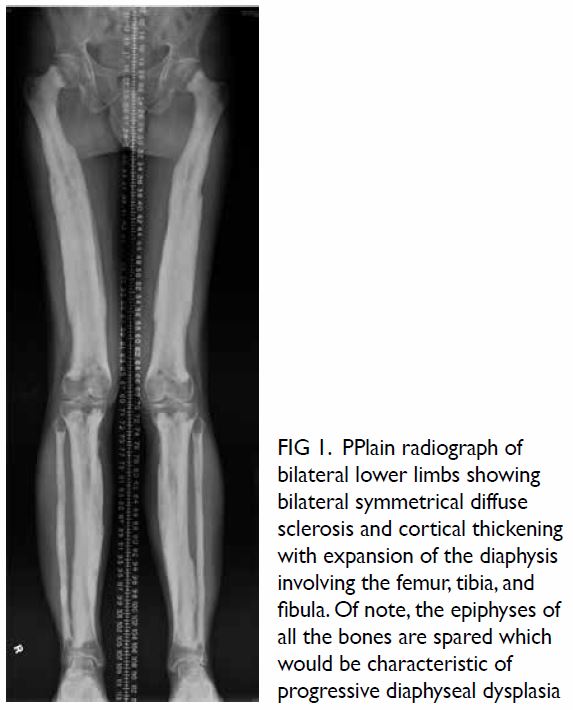
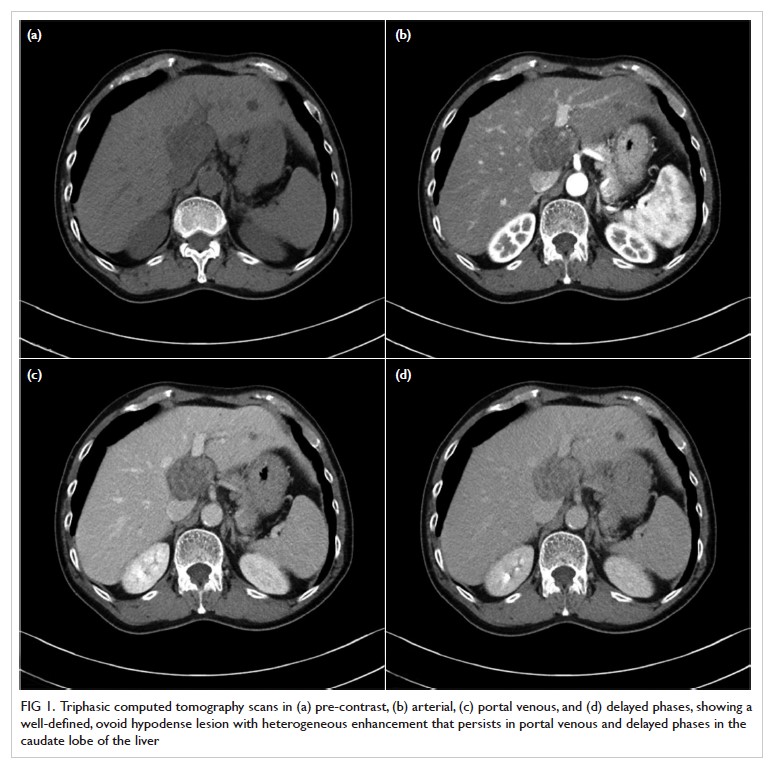
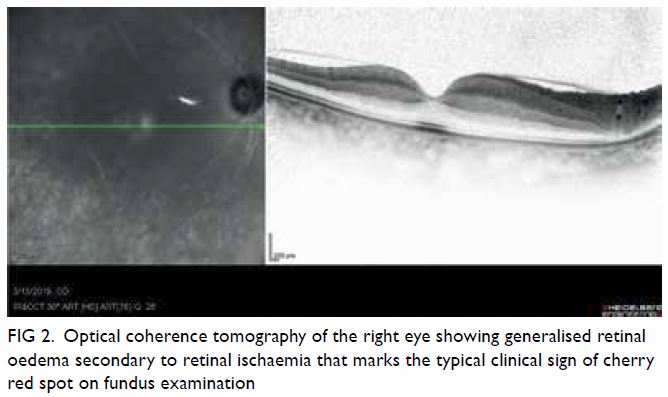
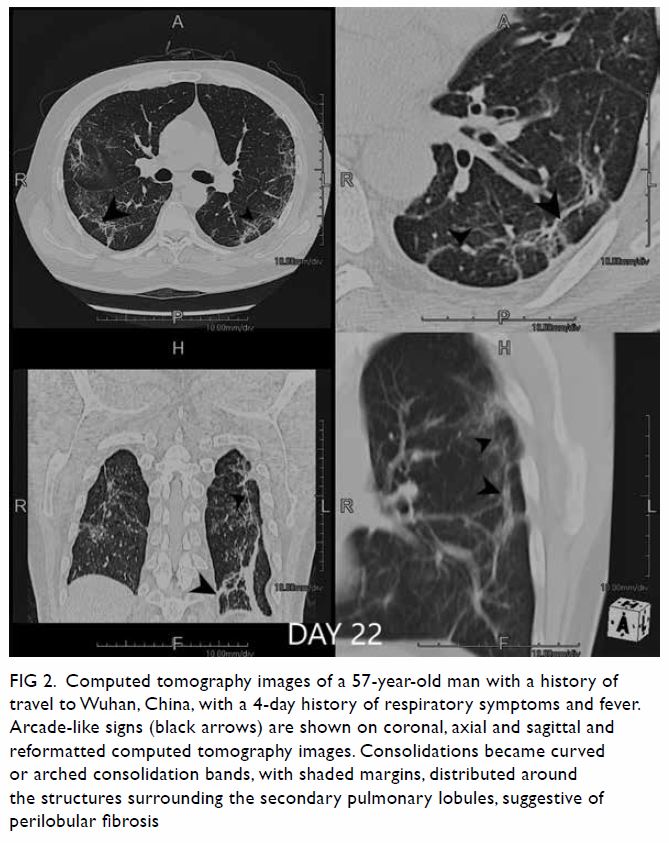
Figure 1. Arterial phase contrast-enhanced computed tomography in (a) coronal and (b) axial planes demonstrating focal renal parenchymal swelling and hypoenhancement (asterisk) with associated perinephric fat stranding (arrow) consistent with acute pyelonephritis. Note the presence of a small renal stone (arrowhead) in a dilated calyx
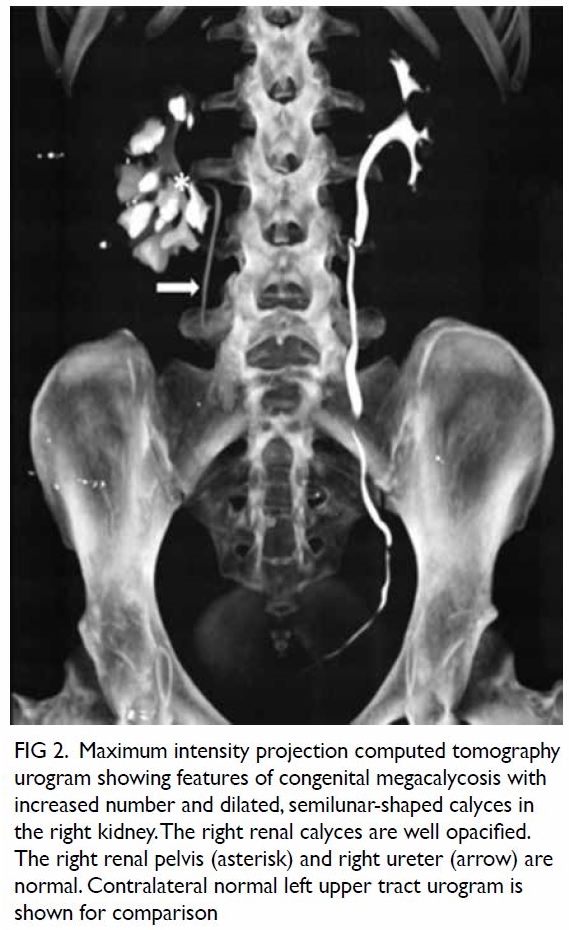
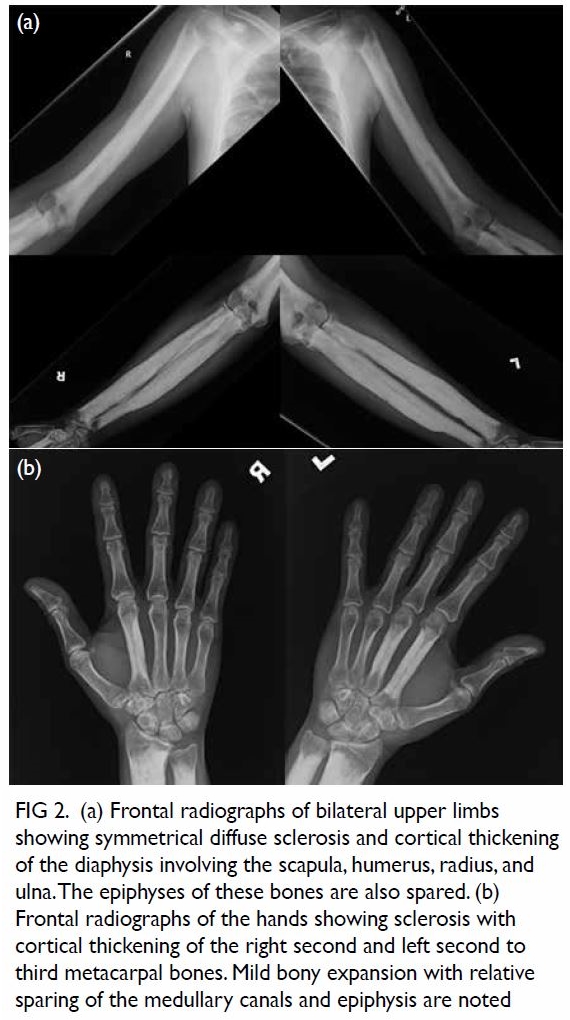
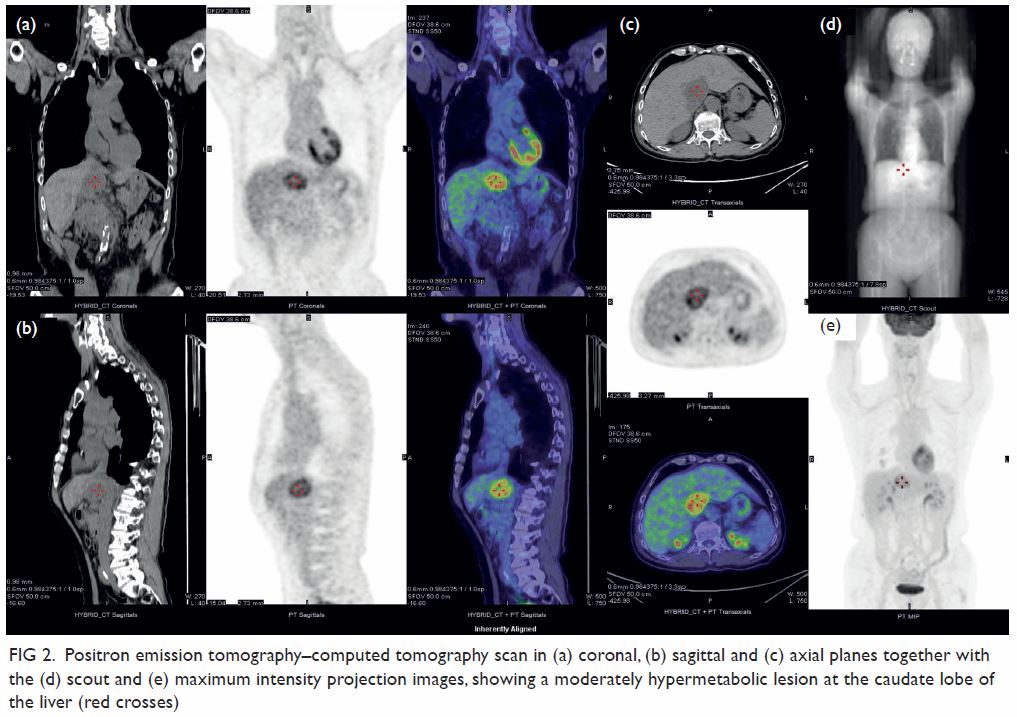
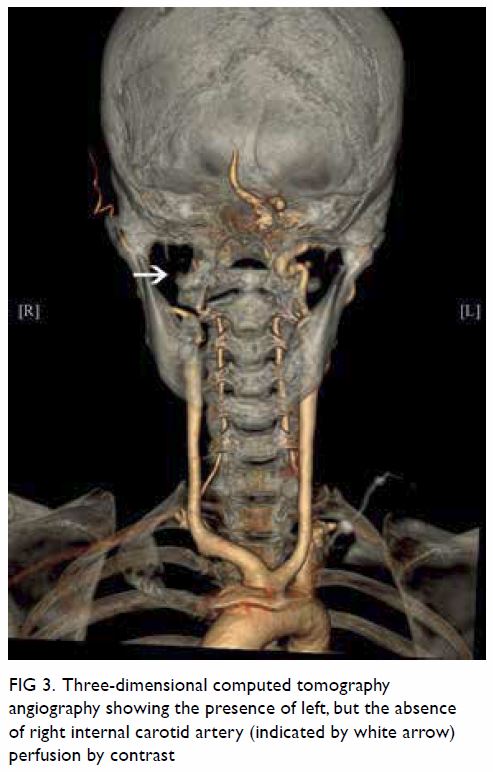
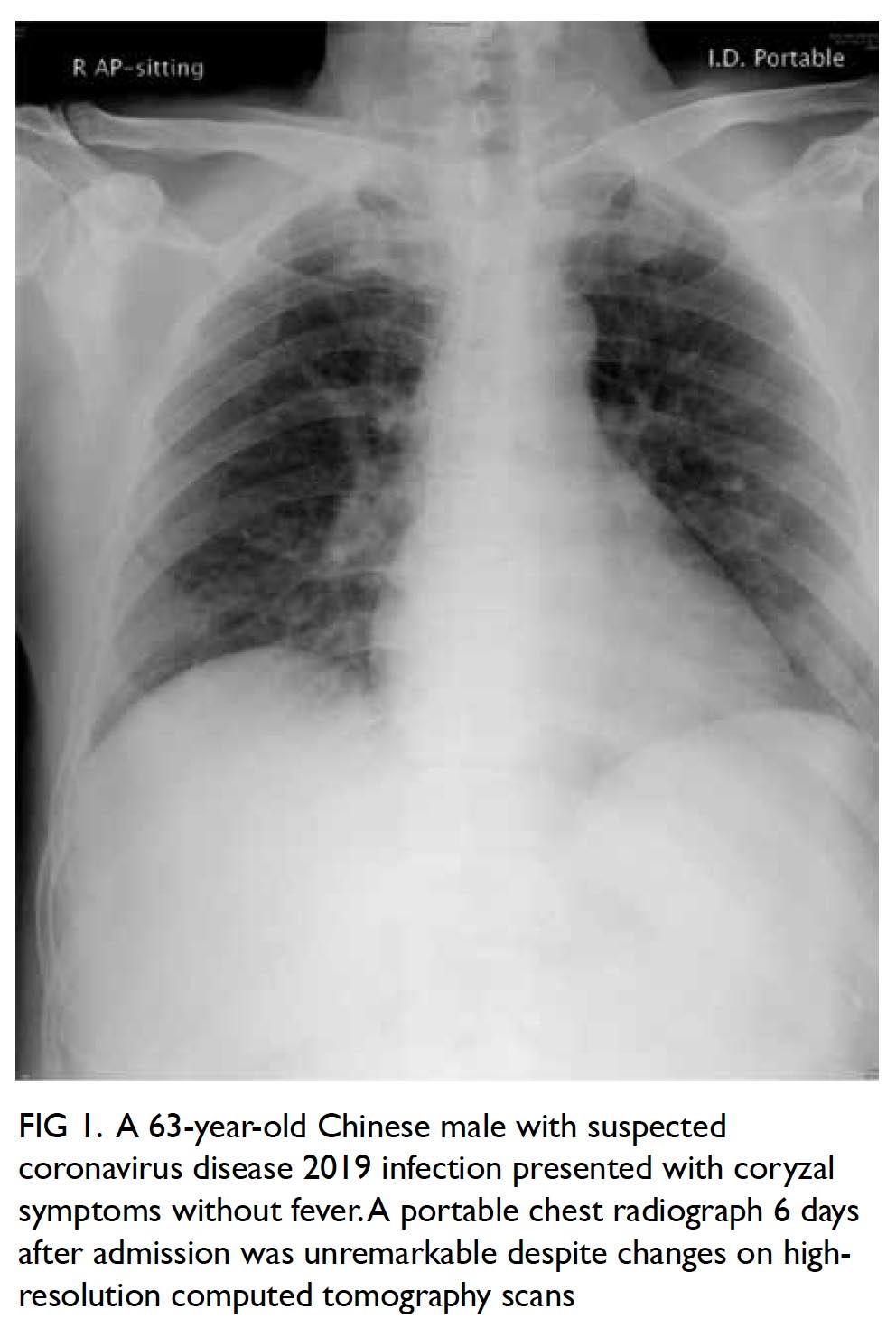
Figure 2. Maximum intensity projection computed tomography urogram showing features of congenital megacalycosis with increased number and dilated, semilunar-shaped calyces in the right kidney. The right renal calyces are well opacified. The right renal pelvis (asterisk) and right ureter (arrow) are normal. Contralateral normal left upper tract urogram is shown for comparison
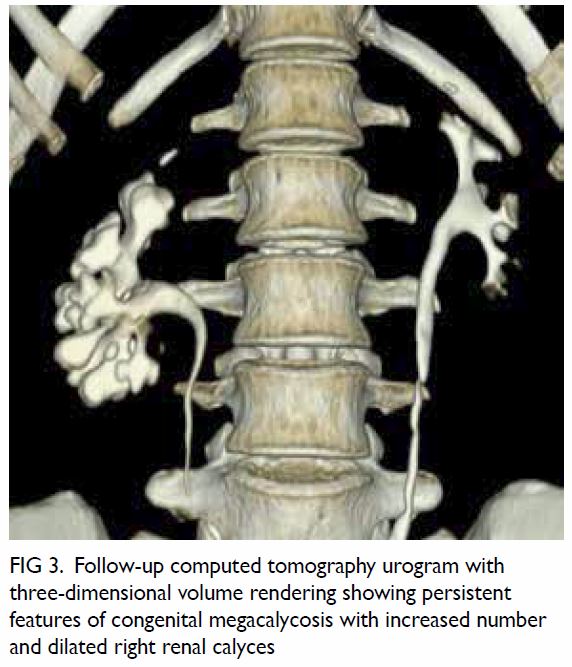
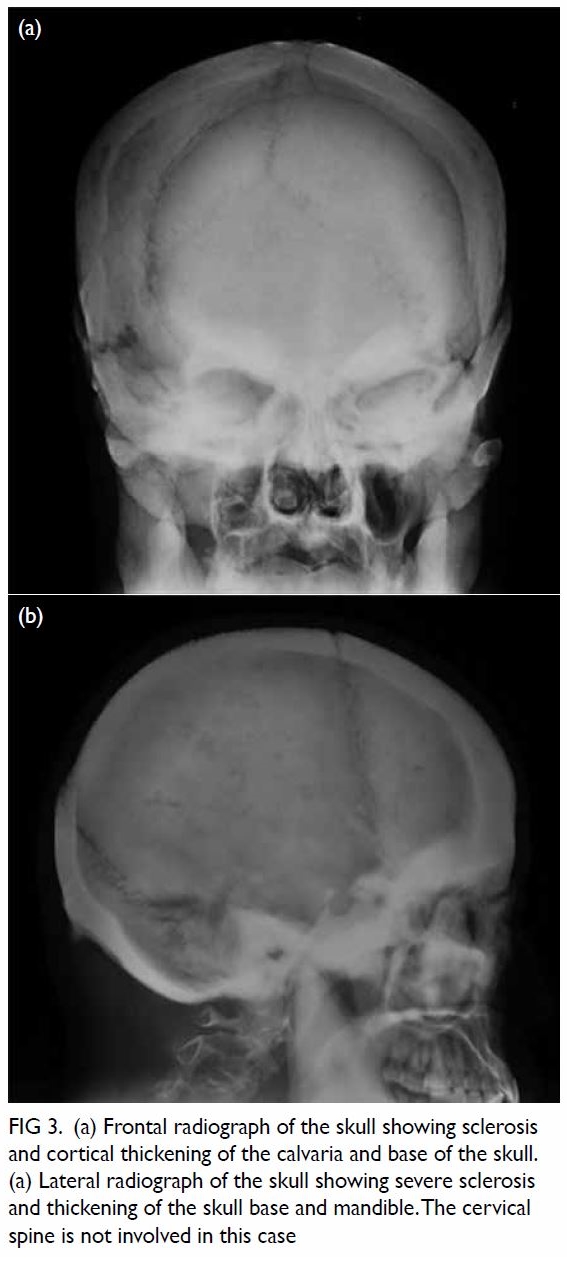
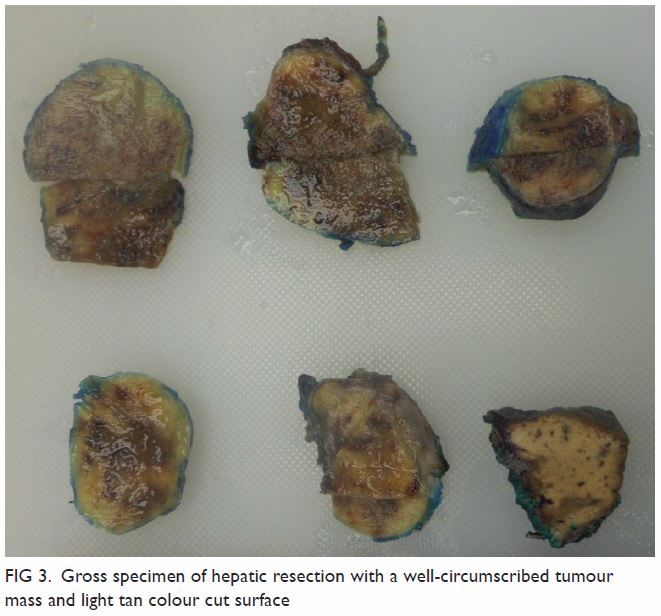
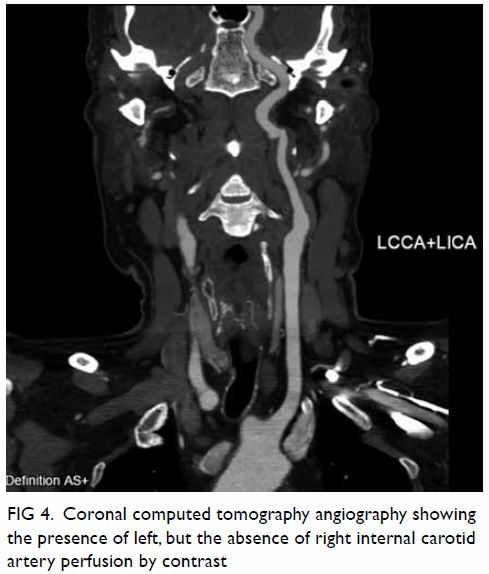
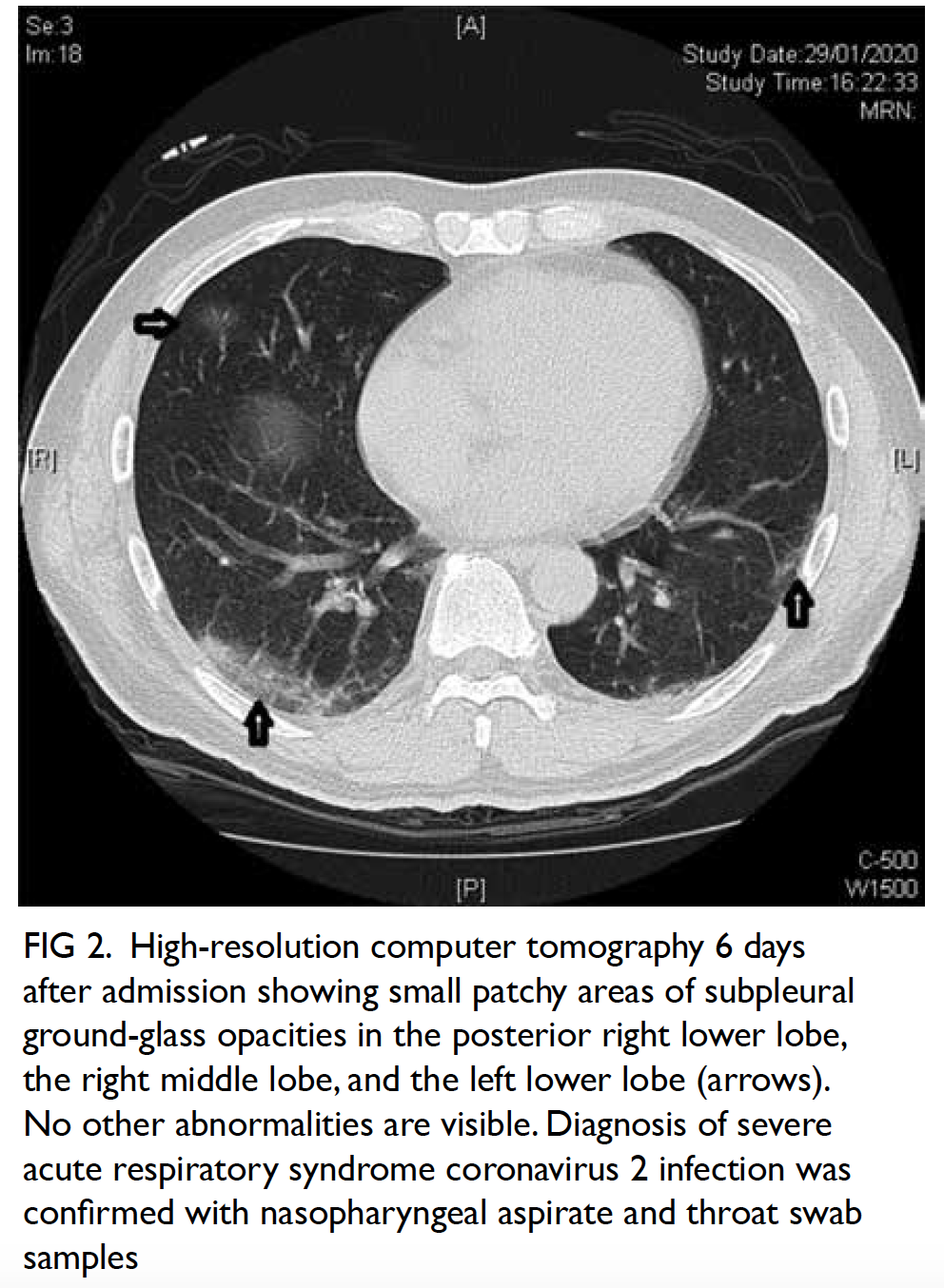
Figure 3. Follow-up computed tomography urogram with three-dimensional volume rendering showing persistent features of congenital megacalycosis with increased number and dilated right renal calyces
The radiological features were diagnostic
of congenital megacalycosis. The anomaly is
characterised by caliectasis with malformation.
The classic triangular or conical shape of the renal
calyces is replaced by a semilunar configuration. The pyramids of Malpighi are hypoplastic and
the tip of each papilla is flat. The calyces have a
rounded appearance with neither fornix nor papillae
impressions. Polycalycosis is another feature of
the condition and typically 20 to 25 calyces can
be identified. The condition is differentiated from
obstruction by the finding of a non-dilated renal
pelvis, infundibulum, and ureter. The renal cortex
was of normal thickness with good concentration
of contrast medium in the distended calyces (Figs 2
and 3).
Congenital megacalycosis is a rare condition
with approximately 100 cases reported in the
literature. The anomaly is found predominantly
in men and usually affects only one kidney. The
exact pathogenesis remains unclear.1 2 3 The condition
is usually asymptomatic and is detected during
workup of urinary stone disease or urinary tract
infection in adults. On the contrary, reports of
congenital megacalycosis in paediatric patients are
on the rise with the increasing use of imaging in
antenatal screening.4 Although urinary stasis in the
distended calyces may predispose to infection and
stone formation,1 2 renal function remains normal
and neither anatomic nor functional deterioration
occur over time.5 The benign nature suggests that
megacalycosis should be considered a condition
rather than a disease, and the term “megacalycose”
has been suggested as an alternative term by some
authors. Treatment should target complications
only. Identification of the condition is important to
avoid unnecessary investigation and intervention in
a normally functioning kidney despite the inherent
anatomic defect.2
Author contributions
Concept or design: CL Cho.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take
responsibility for its accuracy and integrity.
Conflicts of interest
The authors have disclosed no conflicts of interest.
Funding/support
This pictorial medicine paper received no specific grant from
any funding agency in the public, commercial, or not-for-profit
sectors.
Ethics approval
This study was conducted in accordance with the principles
outlined in the Declaration of Helsinki. Verbal consent was
obtained for the purpose of case study.
References
1. Puigvert A. Le megacalice. J Urol Nephrol 1964;70:321-6. Crossref
2. Kimche D, Lask D. Megacalycosis. Urology 1982;19:478-81. Crossref
3. Kalaitzis C, Patris E, Deligeorgiou E, et al. Radiological
findings and the clinical importance of megacalycosis. Res
Rep Urol 2015;7:153-5. Crossref
4. Kasap B, Kavukçu S, Soylu A, et al. Megacalycosis: report of two cases. Pediatr Nephrol 2005;20:828-30. Crossref
5. Redman JF, Neeb AD. Congenital megacalycosis: a
forgotten diagnosis? Urology 2005;65:384-5. Crossref