Differentiating episodic ataxia type 2 from migraine: a case report
Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Differentiating episodic ataxia type 2 from
migraine: a case report
HJ Wu, MRCPCH1; WL Lau, FHKAM (Paediatrics), FHKCPaed1; Tina YC Chan, MB, ChB2; Sammy PL Chen, FHKAM (Pathology), FHKCPath2; CH Ko, FHKAM (Paediatrics), FHKCPaed1
1 Department of Paediatrics, Caritas Medical Centre, Hong Kong
2 Department of Chemical Pathology, Princess Margaret Hospital, Hong Kong
Corresponding author: Dr CH Ko (koch@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Case report
A 17-year-old Chinese male was referred to
Caritas Medical Centre, Hong Kong, in July 2015
for suspected migraine. His parents recalled onset
of symptoms at age 11 years, at which time the
child might suddenly hold onto rails for prolonged
rest while climbing stairs, complaining of marked
dizziness. The child sought no medical advice until
age 17 years, when symptoms worsened to almost
daily attacks. Each episode lasted from >10 minutes
to a few hours and was usually precipitated by exercise
or stress, but not by change in head position, neck
movement, or fasting. During attacks, he could not
walk along a straight line and experienced subjective
generalised weakness. School biannual 9-minute
run stamina assessment was prematurely aborted
due to incapacitating dizziness. The dizziness
was vertigo-like in nature and could be associated
with bilateral temporal headache and nausea. He
reported no tinnitus or dysarthria during attacks.
No abnormal eye movement was observed by his
parents during attacks. There was no history of aura,
photophobia, chest pain, palpitation, numbness,
visual impairment, hearing loss, tinnitus, or
syncope. He functioned normally between attacks.
Family history was negative for recurrent dizziness,
migraine headache, or neurological disease.
Neurological examination results were normal; there
was no gaze-evoked nystagmus, cerebellar signs, or
wide-based gait. Results of systemic examinations
and laboratory tests, including complete blood
picture, electrolytes, and thyroid function, were
unremarkable. Electrocardiogram and computed
tomography of the brain showed no abnormalities.
The patient was initially diagnosed with
migraine variant but experienced no improvement
after a 4-week trial of pizotifen prophylaxis. The
predominantly exercise-induced prolonged vertigo
spells, together with a relative paucity of pulsating
lateralised headaches and lack of response to migraine
treatment prompted the suspicion of episodic ataxia
type 2 (EA2). Acetazolamide was started and the
patient reported rapid clinical improvement with
>50% reduction in frequency and severity of attacks.
The patient was asked to temporarily withhold the medication; symptoms returned instantly but
responded to resumption of acetazolamide. Genetic
testing revealed a novel heterozygous variant
NM_001127222.1 (CACNA1A): c.5067+1 G>A and
was likely to be pathogenic for EA2. This mutation
was not found in the targeted genetic analyses of
the parents and is likely de novo. During follow-up
examinations, the patient reported that he could
complete school 9-minute runs.
Discussion
The most common type of episodic ataxia is EA2
(Table), with an estimated prevalence of <1 in
100 000.1 2 In EA2, the characteristic ataxic spells
can last from >10 minutes to hours. Triggers
include physical exertion, emotional stress,
alcohol, and caffeine. Disease onset is generally
between age 5 and 20 years. Patients are usually
symptom-free between attacks, but there may be
interictal nystagmus and gradual development of
a progressive ataxia syndrome.1 Episodic ataxia
type 2 is caused by mutations in the P/Q-type
voltage-gated calcium channel gene CACNA1A. It
encodes the pore-forming subunit of the P/Q-type
voltage-gated calcium channel, widely expressed
throughout the central nervous system, particularly
on presynaptic terminals of cerebellar Purkinje cells
and granule layer neurones. It plays a key role in
synaptic transmission. More than 80 EA2-associated
mutations on CACNA1A have been reported.1 3
Clinical diagnosis of EA2 is challenging; the
symptoms are often interpreted by primary care
physicians as being due to more common conditions
such as migraine, epilepsy, and vestibular disorders.
Overlapping features with allelic conditions of
familial alternating hemiplegia and spinocerebellar
type 6 may result in phenotype variability
including dysarthria, diplopia, tinnitus, hemiplegia
and headache.1 3 Headache may appear without
accompanying symptoms. The periodic appearance
of ostensibly functional symptoms is often
misinterpreted as migrainous; notably, 50% of EA2
cases fulfil the International Headache Society criteria
for migraine.3 In our patient, differentiating clues
included identification of prolonged ataxic spells, exercise-induced attacks, and a lack of response to
conventional migraine medications. Interictal ataxia
and nystagmus, if present, may also help differentiate
EA2 from migraine. The absence of positional
dizziness and hearing loss helps differentiate EA2
from vestibular disorders.1 Electroencephalogram
monitoring during attacks may help exclude
epilepsy. As illustrated by our patient, dramatic
improvement with acetazolamide may be diagnostic
in doubtful cases. Notwithstanding the therapeutic
response, paroxysmal exercise-induced movement
disorders with acetazolamide responsiveness may
also occur in glucose transporter defects such as
GLUT1 deficiency. It is a rare metabolic defect of
glucose uptake at the blood-brain barrier that may
present as periodic movement disorder. The latter
may have associated features of developmental delay,
microcephaly, and carbohydrate responsiveness that
are not present in EA2. The laboratory hallmark
of GLUT1 deficiency is a low cerebrospinal fluid/blood glucose ratio <0.4.4 Episodic ataxia type 2
is differentiated from other hereditary episodic
ataxias by the age of onset, spell duration, interictal
nystagmus, and genetic locus (Table).1 2 4
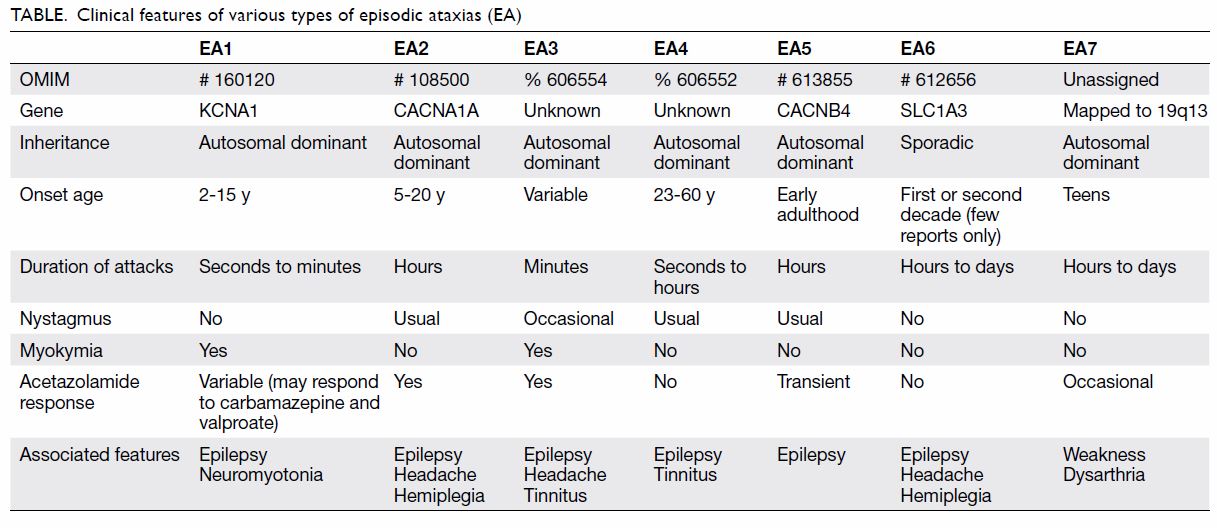
Table. Clinical features of various types of episodic ataxias (EA)
Therapeutically, EA2 has a dramatic response to
acetazolamide, with 50% to 75% of patients reporting
improvement in episode severity and frequency at
doses from 250 to 1000 mg daily.1 Dose escalation
is often limited by side-effects of paraesthesia,
nephrocalcinosis, fatigability and hyperhidrosis.1 5 If
acetazolamide is not tolerated, the potassium channel
blocker 4-aminopyridine may improve symptoms.
The mechanism is not fully understood; in animal
models it has been shown to prolong the action
potentials and restore the diminished precision of
pacemaking in Purkinje cells.5
In summary, EA2 is a rare neurological
disorder that can be misdiagnosed as migraine,
epilepsy, or vestibular disorders, particularly in young children who cannot give a detailed history.
Heightened physician awareness and early treatment
can significantly improve patient quality of life.
Author contributions
All authors contributed to the concept or design of the study,
acquisition of the data, analysis or interpretation of the
data, drafting of the manuscript, and critical revision of the
manuscript for important intellectual content. All authors
had full access to the data, contributed to the study, approved
the final version for publication, and take responsibility for its
accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This case report received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the tenets of the Declaration of Helsinki. The patient consented to publication
of this article in a peer-reviewed journal.
References
1. Guterman EL, Yurgionas B, Nelson AB. Pearls & Oy-sters:
Episodic ataxia type 2 case report and review of the
literature. Neurology 2016;86:239-41. Crossref
2. Jen JC, Graves TD, Hess EJ, et al. Primary episodic ataxias: diagnosis, pathogenesis and treatment. Brain
2007;130:2484-93. Crossref
3. Spillane J, Kullmann DM, Hanna MG. Genetic neurological
channelopathies: molecular genetics and clinical
phenotypes. J Neurol Neurosurg Psychiatry 2016;87:37-48.
4. Kipfer S, Strupp M. The clinical spectrum of autosomal-dominant
episodic ataxias. Mov Disord Clin Pract
2014;1:285-90. Crossref
5. Kalla R, Teufel J, Feil K, Muth C, Strupp M. Update on the pharmacotherapy of cerebellar and central vestibular
disorders. J Neurol 2016;263 Suppl 1:S24-9. Crossref