Necrotising fasciitis: a rare complication of acute appendicitis
DOI: 10.12809/hkmj166180
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Necrotising fasciitis: a rare complication of acute
appendicitis
Thomas WY Chin, MB, ChB; Koel WS Ko, MB, BS; KH
Tsang, MB, BS, FHKAM (Radiology)
Department of Radiology and Imaging, Queen
Elizabeth Hospital, Yaumatei, Hong Kong
Corresponding author: Dr Thomas WY Chin (twychin@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Introduction
Acute appendicitis is a common surgical emergency
that can cause severe complications if diagnosis and management are
delayed.1 Necrotising fasciitis
(NF) is a necrotic infection involving deeper layers of the skin and
subcutaneous tissue that spreads rapidly along the fascia, progressing to
systemic sepsis. It is most commonly induced by injury and is an extremely
rare complication of acute appendicitis. In this article, we present a
case of perforated appendicitis complicated by right thigh and scrotal NF.
Since the condition runs a fulminant clinical course, prompt diagnosis and
early surgical intervention are crucial to reduce mortality.
Case report
A 60-year-old man with good past health presented
to the Accident and Emergency Department of Queen Elizabeth Hospital, Hong
Kong, in December 2015 with a 6-day history of fever and lower abdominal
pain.
His vital signs on presentation were as follows:
systolic blood pressure 97 mm Hg; diastolic blood pressure 59 mm Hg; pulse
90 beats per minute; and body temperature 38.3°C. Physical examination
revealed lower abdominal and right thigh tenderness without subcutaneous
emphysema. Laboratory data showed leucocytosis (15.5 × 109/L)
with neutrophilia (neutrophils 88.7%) and evidence of acute kidney injury
(estimated glomerular filtration rate 38 mL/min/1.73 m2, serum
creatinine 171 μmol/L).
Abdominal, pelvic, and right hip radiographs were
unremarkable. Urgent abdominal and pelvic computed tomographic (CT) scan
revealed a dilated retrocaecal appendix with heterogeneous wall
enhancement, compatible with acute gangrenous appendicitis. There was
rupture at the appendiceal tip, contiguous with a 2.8- × 4.5- × 8.0-cm
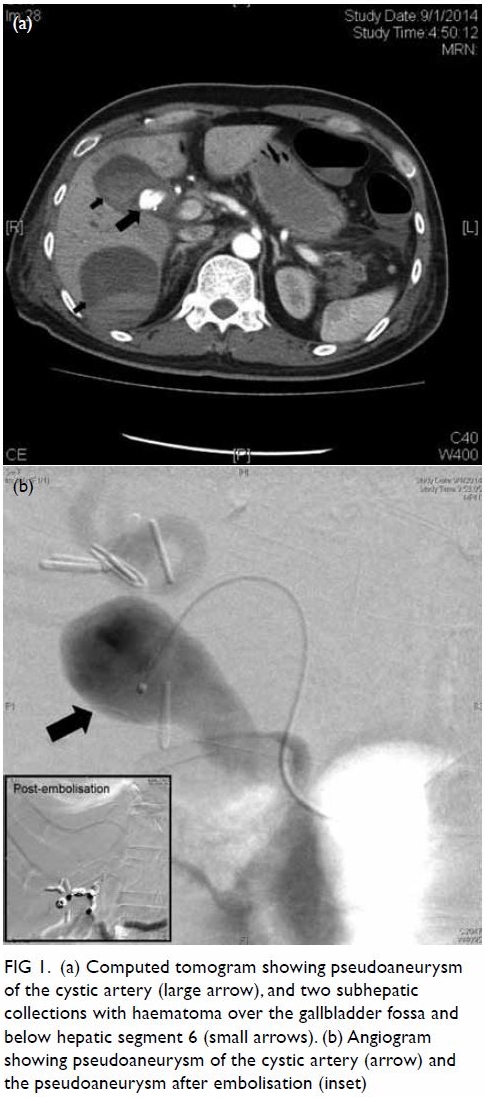
gas-containing abscess posterolateral to the ascending colon and caecum (Fig 1). Increased peri-appendiceal and pericaecal
stranding and free gas were noted. Multiple smaller pelvic abscesses were
also observed. An incidental finding of abnormal swelling of the right
upper thigh was noted in the limited coverage of the thigh in the original
CT scan. Another CT scan including the whole right thigh was performed by
the on-site radiologist. Abnormal high-density fluid and fat stranding
were noted in the subcutaneous layer and intermuscular fascial planes in
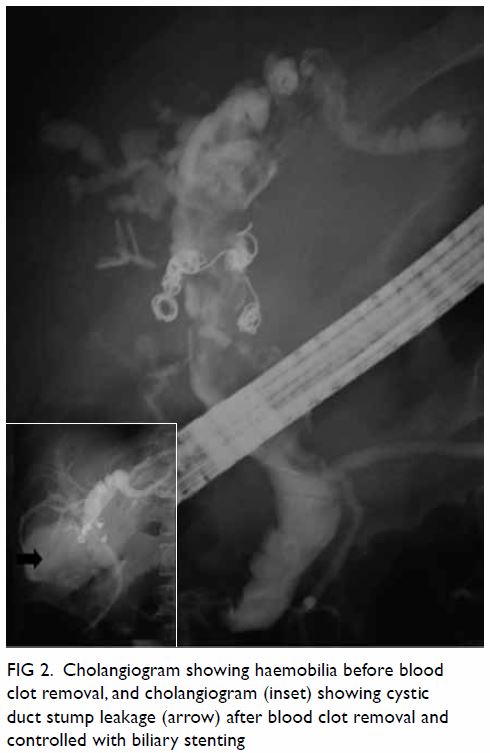
the anterior, adductor, and posterior compartments of the right thigh (Fig 2). The thigh muscles were swollen without
discrete abscess formation. No gas density was detected along the fasciae.
The initial overall imaging diagnosis was ruptured acute appendicitis with
peri-appendiceal abscess, complicated by right thigh NF.
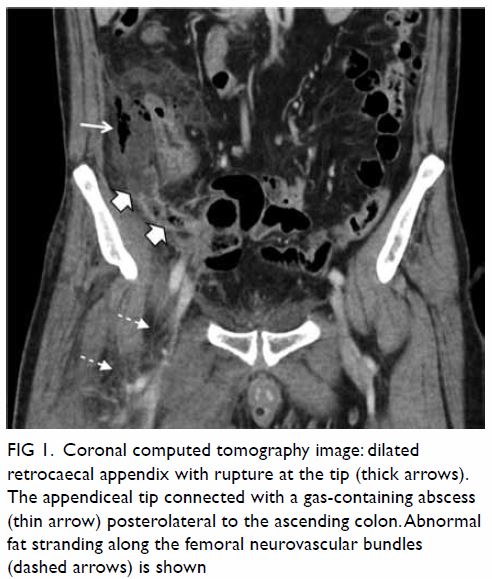
Figure 1. Coronal computed tomography image: dilated retrocaecal appendix with rupture at the tip (thick arrows). The appendiceal tip connected with a gas-containing abscess (thin arrow) posterolateral to the ascending colon. Abnormal fat stranding along the femoral neurovascular bundles (dashed arrows) is shown
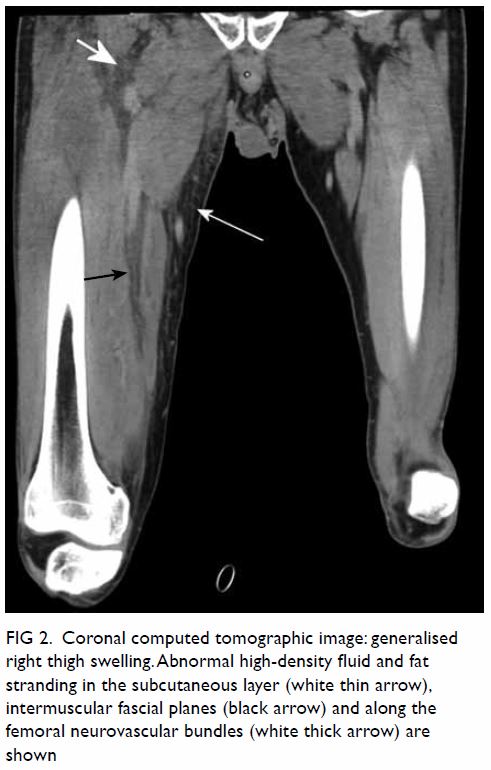
Figure 2. Coronal computed tomographic image: generalised right thigh swelling. Abnormal high-density fluid and fat stranding in the subcutaneous layer (white thin arrow), intermuscular fascial planes (black arrow) and along the femoral neurovascular bundles (white thick arrow) are shown
Subsequent urgent laparotomy revealed a ruptured
inflamed retrocaecal appendix and an 8-cm retrocolic abscess. Appendectomy
and abscess drainage were performed. These were followed by exploration of
the right thigh that revealed oedematous and necrotic subcutaneous and
deep soft tissues mainly involving the adductor compartment. Multiple
intramuscular abscesses were seen with turbid fluid tracing along the
femoral neurovascular bundles proximally to the pelvic region. Overall
features were compatible with NF. Right above-knee amputation with
excisional debridement was performed in view of the patient’s poor general
condition and extensive involvement.
Culture of the debrided muscle and fascial tissue
yielded Escherichia coli, Bacteroides fragilis, and Streptococcus
milleri. A combination of antibiotics was given, including amikacin,
ampicillin, levofloxacin, linezolid, meropenem, and penicillin G. However,
septic shock persisted with development of multi-organ dysfunction despite
multiple inotropes. Another CT scan of the abdomen and pelvis was
performed on postoperative day 1, revealing a grossly swollen scrotum with
hydrocoele (not shown). No intrascrotal gas density was detected. In view
of the patient’s clinical deterioration, urgent scrotal exploration was
performed and revealed extensive scrotal skin necrosis with copious serous
fluid draining from the perineum.
After the second surgery, the patient had
persistent multi-organ failure with clinical deterioration despite
supportive management. He developed acute respiratory distress syndrome
with respiratory failure and disseminated intravascular coagulation with
severe anaemia, further complicated by acute coronary syndrome. The
patient succumbed on the fourth day of hospital admission.
Discussion
The incidence of NF has risen recently owing to an
increased prevalence of patients who are immunocompromised secondary to
diabetes mellitus or treatment for malignancy or human immunodeficiency
virus infection.1 The mortality
rate for NF is high, despite significant advances in antibiotic treatment,
owing to its rapid progression to septic shock and multi-organ failure.1
Common causes of NF include minor trauma, skin
infection, intravenous drug use, and surgical complication.1 The abdomen,
groin, and extremities are the regions most commonly affected by NF.1 2 3 4 5 Necrotising fasciitis is commonly polymicrobial, caused
by virulent toxin-producing bacteria such as Group A haemolytic
streptococci and Staphylococcus aureus. Other causative bacteria
include Bacteroides, Clostridium, Enterobacteriaceae,
Peptostreptococcus, Proteus, Pseudomonas, and Klebsiella.1
The diagnosis of NF is challenging due to its
rarity and non-specific presentation. Usually the early manifestations are
local inflammatory signs and symptoms such as swelling, erythema, and
tenderness, occasionally with fever. However, NF should be suspected when
the severity of pain or clinical status are disproportional to local
findings.5 Laboratory studies show
leucocytosis with predominant neutrophilia,1
but is unfortunately non-specific.5
Presence of soft tissue gas in the absence of penetrating trauma suggests
a diagnosis of NF,2 although the absence of soft tissue gas does not
exclude the diagnosis, as evidenced by our case.
Plain radiographs are usually unhelpful for initial
diagnosis as soft tissue gas is seldom detected until late in the disease
process.1 Magnetic resonance
imaging is superior in delineating soft tissue pathology but is a
suboptimal modality for critically ill patients.2
Computed tomography is the preferred imaging modality, with reported
findings including asymmetric fascial thickening with fat stranding and
subcutaneous gas tracking along fascial planes.2
It also delineates the extent of tissue involvement well and is useful for
monitoring treatment response.
Necrotising fasciitis secondary to perforated
appendicitis is rarely reported. We have found only 18 case reports of NF
caused by acute appendicitis published in the English literature.1 2 3 4 5 The affected regions included the abdominal wall (most
commonly involved), perineum, and thigh. According to Taif and Alrawi,5 there have been only three reported cases of
appendicitis complicated by NF predominantly involving the lower limb. All
cases involved the right thigh, likely from more direct infective spread
along the right femoral neurovascular bundles than the left.
Our case represents a rare but life-threatening
consequence of a common disease despite the absence of risk factors. Early
clinical changes of NF can be subtle. Early recognition, broad-spectrum
antibiotic treatment, and aggressive surgical debridement are the
cornerstones of management for this potentially lethal disease.1 A high index of suspicion is needed in diagnosis such
that emergent surgical intervention can be initiated to improve clinical
outcome.
Author contributions
All authors contributed to the design, acquisition
and interpretation of data, drafting of the article, and critical revision
for important intellectual content.
Acknowledgement
We would like to thank the staff radiographers of
the Department of Radiology and Imaging, Queen Elizabeth Hospital for
their assistance in acquisition of the images.
Declaration
All authors have disclosed no conflicts of
interest. All authors had full access to the data, contributed to the
study, approved the final version for publication, and take responsibility
for its accuracy and integrity.
References
1. Chen CW, Hsiao CW, Wu CC, Jao SW, Lee
TY, Kang JC. Necrotizing fasciitis due to acute perforated appendicitis:
case report. J Emerg Med 2010;39:178-80. Crossref
2. Hung YC, Yang FS. Necrotizing
fasciitis—a rare but severe complication of perforated appendicitis.
Radiol Infect Dis 2015;2:81-3. Crossref
3. Hua J, Yao L, He ZG, Xu B, Song ZS.
Necrotizing fasciitis caused by perforated appendicitis: a case report.
Int J Clin Exp Pathol 2015;8:3334-8.
4. Wanis M, Nafie S, Mellon JK. A case of
Fournier’s gangrene in a young immunocompetent male patient resulting from
a delayed diagnosis of appendicitis. J Surg Case Rep 2016;2016:rjw058. Crossref
5. Taif S, Alrawi A. Missed acute
appendicitis presenting as necrotising fasciitis of the thigh. BMJ Case
Rep 2014;2014:bcr2014204247. Crossref