Gas embolism and hyperbaric oxygen therapy: a case series
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Gas embolism and hyperbaric oxygen therapy:
a case series
Jeffrey CW Chau, MB, BS, MRCS(Ed)1; Joe KS Leung, MB, ChB, FRCS1; WW Yan, FRCP, FHKCP2
1 Department of Accident and Emergency, Pamela Youde Nethersole Eastern Hospital, Hong Kong
2 Intensive Care Unit, Pamela Youde Nethersole Eastern Hospital, Hong Kong
Corresponding author: Dr Jeffrey CW Chau (drjhealthcare@gmail.com)
 Full paper in PDF
Full paper in PDF
Case report
Patient 1
A 79-year-old man presented with left-sided
weakness, drowsiness and dysphasia following left
atrial appendage occlusion implantation in a cardiac
intensive care unit. A sudden drop in systolic blood
pressure to 40 mmHg was noted. Echocardiogram
and angiogram revealed no obvious coronary artery
disease. Computed tomography (CT) of the brain
showed no major vessel occlusion or gas pocket. A
new CT of the brain showed more established infarct
and oedema. Four sessions of hyperbaric oxygen
therapy (HBOT) were conducted in accordance with
United States Navy Treatment Tables (USNTT)1: two USNTT6 and two USNTT5.
Patient 2
A 40-year-old woman was admitted to a surgical
unit for suspected acute appendicitis. Urgent CT
of the abdomen with contrast was performed, but
approximately 60 mL of air was injected intravenously
during the procedure and gas was evident in the
right ventricle. Luckily, she was asymptomatic as the
gas emboli had entered only the venous system and
remained in the heart. One session of HBOT was
conducted with USNTT6.
Patient 3
A 57-year-old woman underwent fine needle
aspiration cytology under CT guidance for lung
mass. The patient developed sudden onset bilateral
anopia and left upper limb weakness during the
procedure. Computed tomography brain and thorax
identified a small gas embolism in the pulmonary
vein with pulmonary haemorrhage. No obvious gas
bubble was noted in the brain. A total of four sessions
of HBOT were conducted with two USNTT6 and
two USNTT5.
Patient 4
An 84-year-old man underwent CT-guided lung
biopsy for right lung mass but experienced an acute
deterioration in Glasgow Coma Scale (GCS) score. Urgent CT of the brain showed multiple acute gas
emboli and confirmed the diagnosis of cerebral
arterial gas embolism (AGE). On arrival at the HBOT
centre, he had GCS of 13/15 and right limb weakness
with power of 1/5 and 3/5, respectively. Two sessions
of HBOT were conducted with USNTT6.
Patient 5
A 66-year-old man with biliary pancreatitis
underwent urgent endoscopic retrograde
cholangiopancreatography. After the procedure he
developed left-sided weakness with limb power of
0/5 after 5 minutes. Urgent CT of the brain revealed
branching gas densities at the sulcal space of his
right frontal and parietal lobes (Fig 1). Two sessions
of HBOT with USNTT6 were conducted and the
patient’s symptoms considerably improved.
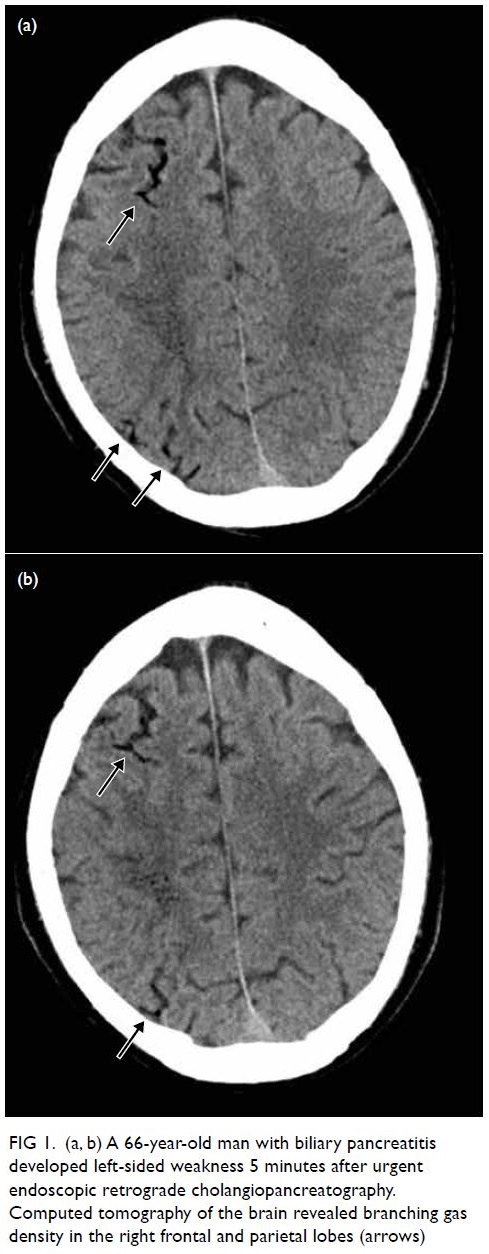
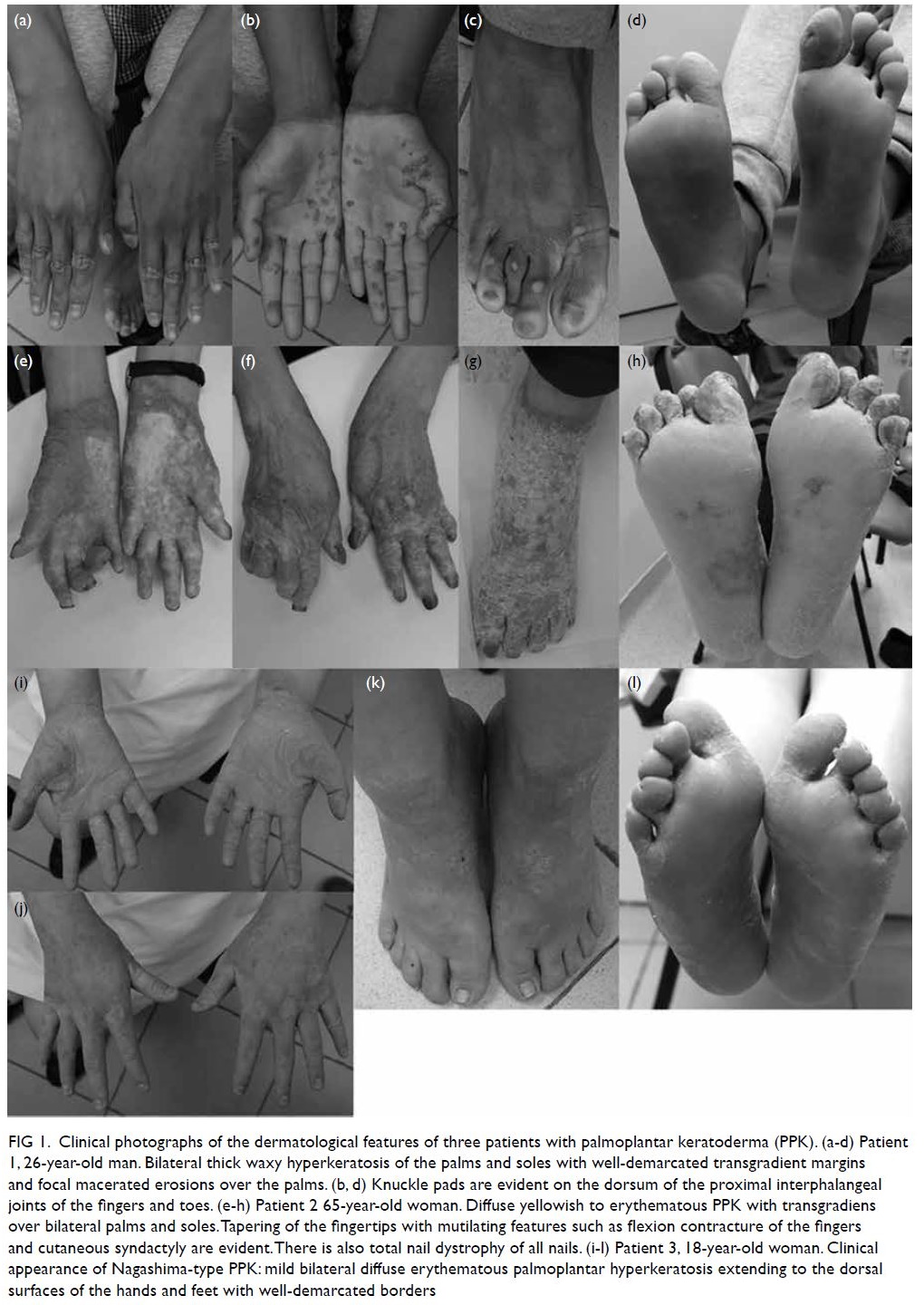
Figure 1. (a, b) A 66-year-old man with biliary pancreatitis developed left-sided weakness 5 minutes after urgent endoscopic retrograde cholangiopancreatography. Computed tomography of the brain revealed branching gas density in the right frontal and parietal lobes (arrows)
Patient 6
A 72-year-old man underwent CT-guided fine
needle aspiration cytology for a lung lesion. The
patient developed transient hypotension on
needle removal and CT of the thorax revealed gas
embolism in his left ventricle with no pneumothorax
(Fig 2). The patient remained asymptomatic with
no neurological deficit. Gas embolism had been
introduced through the pulmonary circulation but
remained in the heart, avoiding the complication of
cerebral arterial occlusion. One session of HBOT
was conducted with USNTT6.
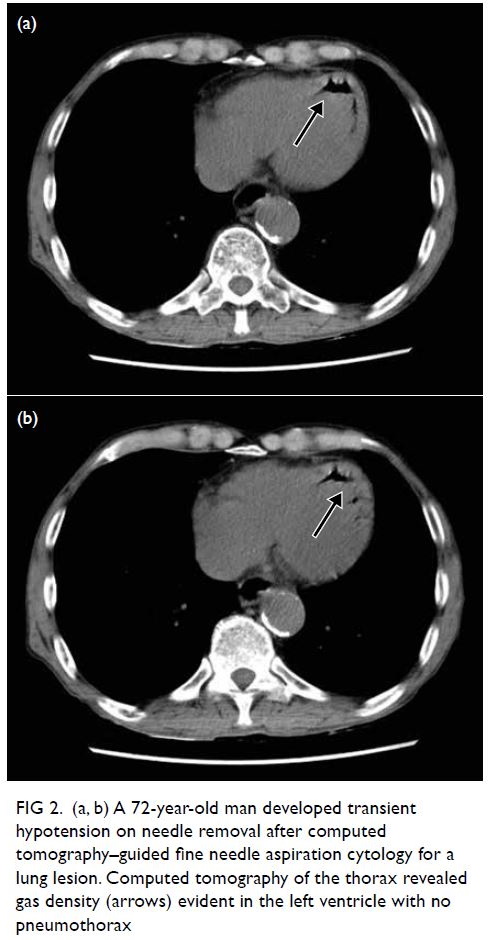
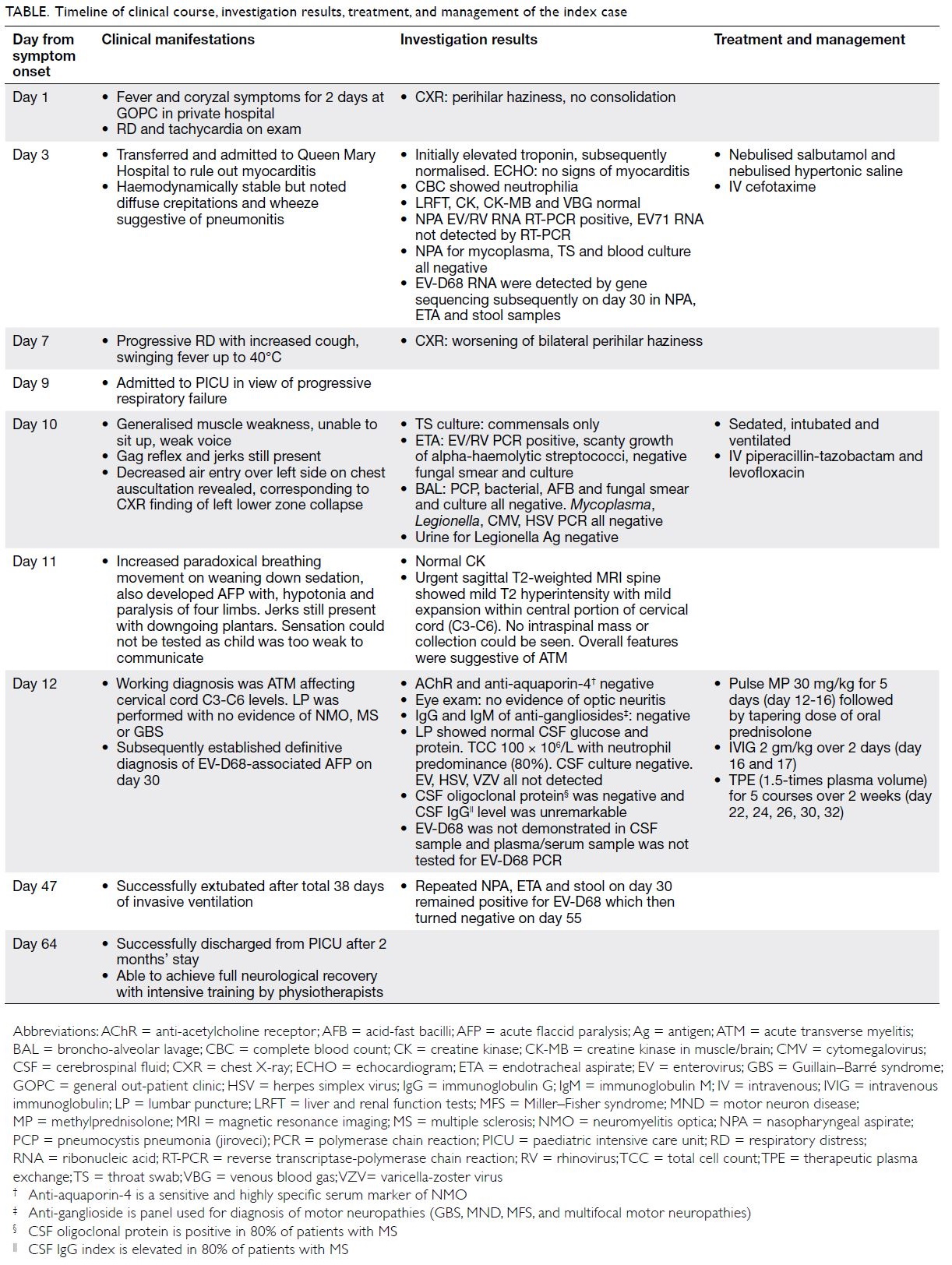
Figure 2. (a, b) A 72-year-old man developed transient hypotension on needle removal after computed tomography–guided fine needle aspiration cytology for a lung lesion. Computed tomography of the thorax revealed gas density (arrows) evident in the left ventricle with no pneumothorax
Discussion
Gas embolism is a serious and life-threatening
condition. It is usually iatrogenic and should be
treated promptly to prevent serious complications
such as cerebral AGE. It presents with sudden
deterioration in neurological and haemodynamic
status following risky medical procedures, most
notably lung biopsy and cardiac interventions. This
case report revealed six patients with gas embolism:
five with AGE and one with venous gas embolism.
Four patients with AGE manifested as cerebral AGE
with neurological deficits such as limb weakness and
anopia (Table).
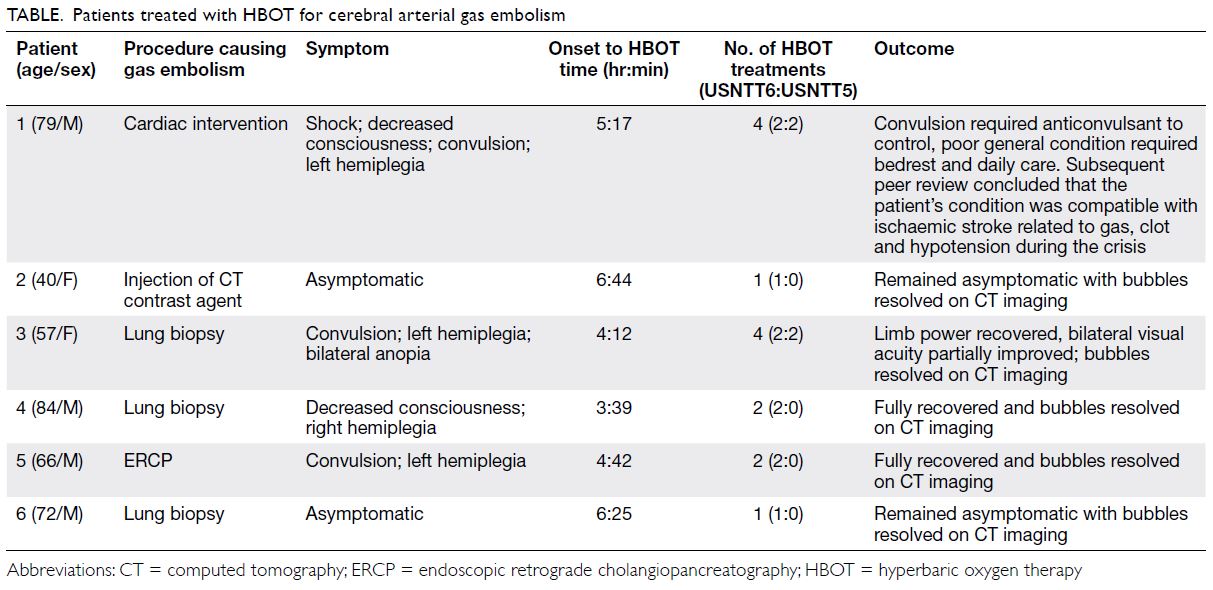
Table. Patients treated with HBOT for cerebral arterial gas embolism
Rationale for using hyperbaric oxygen
therapy
As soon as AGE is suspected, the patient should
start receiving 100% high-flow oxygen and be
placed in the right lateral decubitus position. The
definitive treatment for AGE is HBOT with 100%
oxygen, although no randomised controlled trials have been conducted to confirm its efficacy in this
disease. Hyperbaric oxygen therapy “crushes” the
bubbles with pressure, accelerates gas diffusion
and bubble resolution with oxygen, oxygenates
ischaemic tissue, reduces cerebral oedema, and
decreases neutrophil adhesion to the endothelium.1 2
A retrospective study in France showed that the
initial neurological symptoms were impaired
consciousness (70%), focal motor deficits (60%),
seizures (11%), visual disturbance (10%), dysarthria/aphasia (5%) and vertigo (5%). In all, 26% of patients
were haemodynamically unstable and subject to
hypotension, circulatory collapse, and cardiac arrest.
Respiratory disturbances such as dyspnoea, cough or
chest pain were present in 23% of patients.3
In a study of 45 patients with AGE, good
neurological outcome was achieved in 27 (60%)
of them.4 Time to receipt of HBOT was the only
statistically significant factor predictive of a good outcome with mean delay 8.8 hours.4 Although
probability of a good outcome is highest when HBOT
is administered as soon as possible, a good response
can still be obtained if treatment is delayed for longer
than 24 hours.3 5 There is a tendency for patients
with AGE to deteriorate after their initial apparent
recovery; thus, early HBOT is still recommended
for patients with a seemingly spontaneous recovery.6
Although identification of gas bubbles on CT of the
brain is not a prerequisite to HBOT, early treatment
can also attenuate leucocyte adherence to damaged
endothelium and secondary inflammation that in
turn facilitate the return of blood flow. However, gas
bubbles can persist for several days, and many case
reports have demonstrated the benefit of HBOT
after delays ranging from hours to days. Complete
recovery has been reported in one patient in whom
treatment was delayed for 28 hours. In 2017,
another patient who was deeply comatose with AGE
recovered and was able to lead a functional life after
HBOT had been delayed for 6 days.7 8 In our cases,
all the patients were treated within 7 hours onset of
symptoms. Most fully recovered except for Patient 1.
The aetiology of neurological deterioration in
Patient 1 was likely due to a combination of gas
embolism, clots, and hypotension.
Hyperbaric oxygen therapy treatment tables
According to the United States Navy Diving Manual,9
USNTT6 is the standard initial compression therapy
for AGE. It is a table with maximum pressure up to
2.8 atmospheres absolute (ATA) for 1 hour 15 minutes
with slow depressurisation to 1.9 ATA lasting for
2.5 hours. Total treatment time is approximately 4 hours 45 minutes. There is no conclusive evidence
that use of pressure higher than 2.8 ATA has any
advantage. Use of pressure up to 6 ATA poses further
risks to the patient and attending staff, including gas
toxicity and decompression sickness. Typically, one
to three hyperbaric treatments are required to treat
AGE; the clinical condition should be monitored
until there is no further stepwise improvement.
There are reports that up to 10 treatments have been
required to reach a stable condition.10 11
The USNTT5, which is a shorter treatment
used to treat mild decompression sickness, may
be used together with USNTT6 for AGE when
prolonged treatment is required. This treatment
table advises a maximum pressure up to 2.8 ATA for
45 minutes with slow depressurisation to 1.9 ATA for
30 minutes. Total treatment time is approximately
2 hours 15 minutes. According to the Royal Navy
Diving manual,12 which is commonly used in Europe,
RN62 Table is the standard to treat AGE. This
treatment is similar to USNTT6 with a maximum
pressure of 2.8 ATA.
Conclusion
Arterial gas embolism presents in highly variable ways with no pathognomonic signs or symptoms. It
is uncommon to achieve complete consensus on the
diagnosis, and it is common practice to err on the
side of caution. Hyperbaric oxygen therapy should
be initiated as early as possible although delayed
treatment is also effective. Failure to administer
treatment can result in long-term disability.
Hyperbaric oxygen therapy should be considered for
any suspected AGE.
Author contributions
Concept or design: JCW Chau.
Acquisition of data: JCW Chau.
Analysis or interpretation of data: JCW Chau.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: JCW Chau.
Analysis or interpretation of data: JCW Chau.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patients were treated in accordance with the Declaration of Helsinki. The patients provided informed consent for the
treatment/procedures, and verbal consent for publication.
References
1. United States Navy. Diagnosis and treatment of
decompression sickness and arterial gas embolism. In: US
Navy Diving Manual. Revision 7. Vol 5. Available from:
https://www.navsea.navy.mil/Portals/103/Documents/SUPSALV/Diving/US%20DIVING%20MANUAL_REV7.pdf?ver=2017-01-11-102354-393. Accessed 30 Sep 2020.
2. Grim PS, Gottlieb LJ, Boddie A, Batson E. Hyperbaric oxygen therapy. JAMA 1990;263:2216-20. Crossref
3. Blanc P, Boussuges A, Henriette K, Sainty JM, Deleflie M. Iatrogenic cerebral air embolism: importance of an early hyperbaric oxygenation. Intensive Care Med 2002;28:559-63.Crossref
4. Beevor H, Frawley G. Iatrogenic cerebral gas embolism: analysis of the presentation, management and outcomes of patients referred to The Alfred Hospital Hyperbaric Unit.
Diving Hyperb Med 2016;46:15-21.
5. Massey EW, Moon RE, Shelton D, Camporesi EM. Hyperbaric oxygen therapy of iatrogenic air embolism. J
Hyperb Med 1990;5:15-21.
6. Pearson RR, Goad RF. Delayed cerebral edema complicating cerebral arterial gas embolism: case histories. Undersea Biomed Res 1982;9:283-96.
7. Benson J, Adkinson C, Colier R. Hyperbaric oxygen therapy of iatrogenic cerebral arterial gas embolism. Undersea Hyperb Med 2003;30:117-26.
8. Perez MF, Ongkeko Perez JV, Serrano AR, Andal MP, Aldover MC. Delayed hyperbaric intervention in lifethreatening decompression illness. Diving Hyperb Med
2017;47:257-9 Crossref
9. United States Navy. US Navy Diving Manual. Revision 7.
Vol 5: Diving medicine and recompression chamber
operations. 2016. Available from: https://www.navsea.navy.mil/Portals/103/Documents/SUPSALV/Diving/US%20DIVING%20MANUAL_REV7.pdf?ver=2017-01-11-102354-393. Accessed 30 Sep 2020.
10. Vann RD, Butler FK, Mitchell SJ, Moon RE. Decompression illness. Lancet 2011;377:153-64.Crossref
11. Undersea & Hyperbaric Medical Society. UHMA Best
Practice Guidelines: Prevention and Treatment of
Decompression sickness and arterial gas embolism.
Available from: https://www.uhms.org/images/DCS-AGE-Committee/dcsandage_prevandmgt_uhms-fi.pdf. Accessed 30 Sep 2020.
12. Royal Navy, Ministry of Defence. Diving Manual. HM Stationery Office, London; 1972.