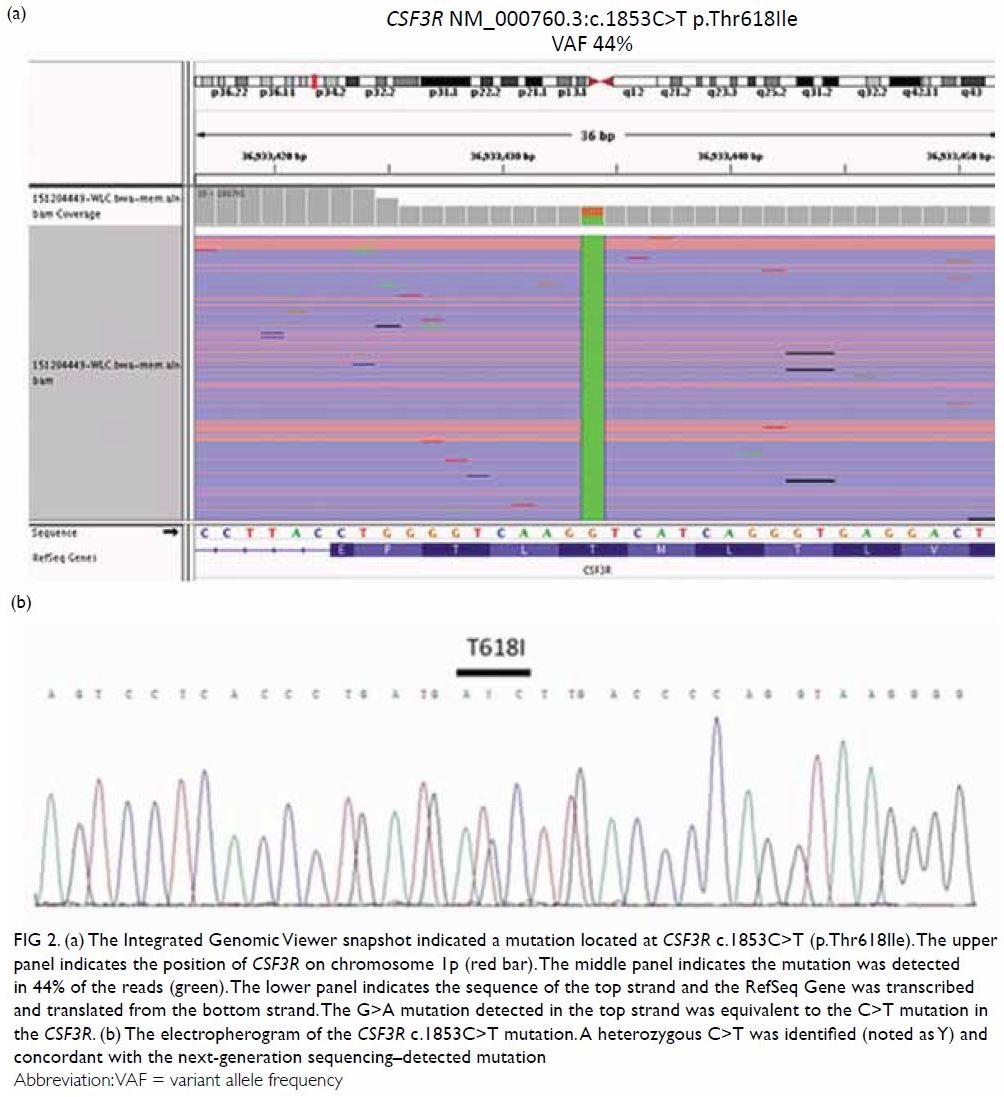
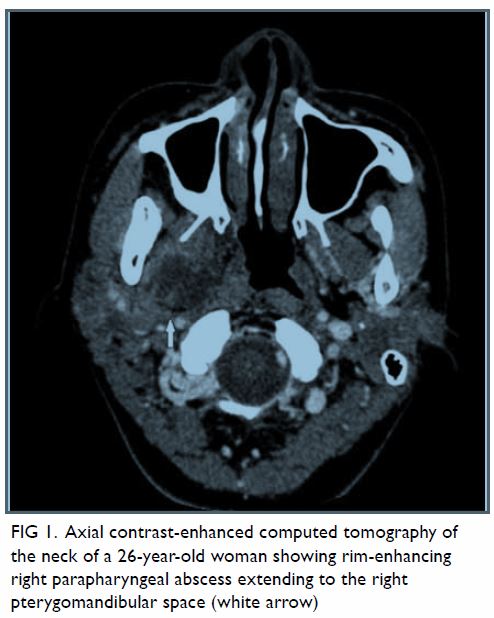
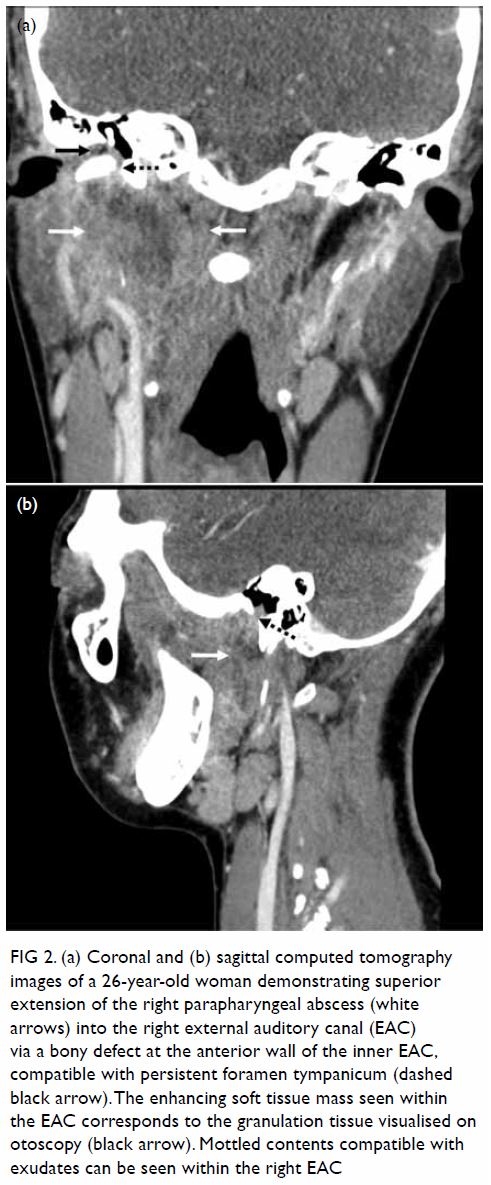
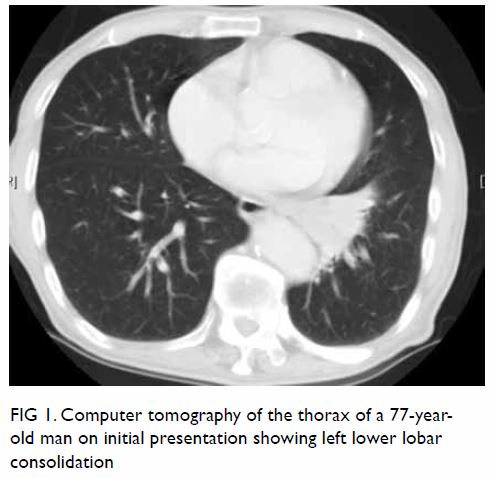
Persistent hypoglossal artery with a contralateral hypoglossal canal venous lake: a case report
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Persistent hypoglossal artery with a contralateral
hypoglossal canal venous lake: a case report
Nuno Vaz, MD1; WL Poon, FRCR, FHKCR2;
SS Cheng, FRCR, FHKCR2
1 Center for Diagnostic Imaging,
Barcelona Clinic Hospital, Barcelona, Spain
2 Department of Radiology and Imaging,
Queen Elizabeth Hospital, Jordan, Hong Kong
Corresponding author: Dr WL Poon (poonwl@ha.org.hk)
 Full
paper in PDF
Full
paper in PDF
Case report
A 44-year-old woman presented to the
emergency department in December 2014 with acute severe
right-sided headache that began at the occipital region and spread to the
right temporal and frontal regions. Pain was only partially relieved by
analgesics. The patient had no history of altered mental state or focal
neurological deficits. Her medical history was unremarkable except for a
road traffic accident a few months previously ago with consequent right
lower limb trauma. Neurological assessment revealed no gross abnormality.
An urgent non-contrast brain computed tomography (CT) scan showed no
intracranial haemorrhage or other abnormalities. Due to persistence of
symptoms the patient underwent brain magnetic resonance (MR) imaging at a
private centre. A small skull lesion was evident on the right basiocciput
for which further imaging study was requested at our hospital. A contrast
3-T MR scan with angiography sequences revealed that the previously
reported lesion corresponded to a 0.7-cm T1-weighted isointense and
T2-weighted hyperintense structure located at the right hypoglossal canal,
which was expanded. It exhibited intense contrast enhancement and was in
direct continuity with the inferior petrosal sinus and the internal venous
plexus around the foramen magnum, all findings suggestive of a venous lake
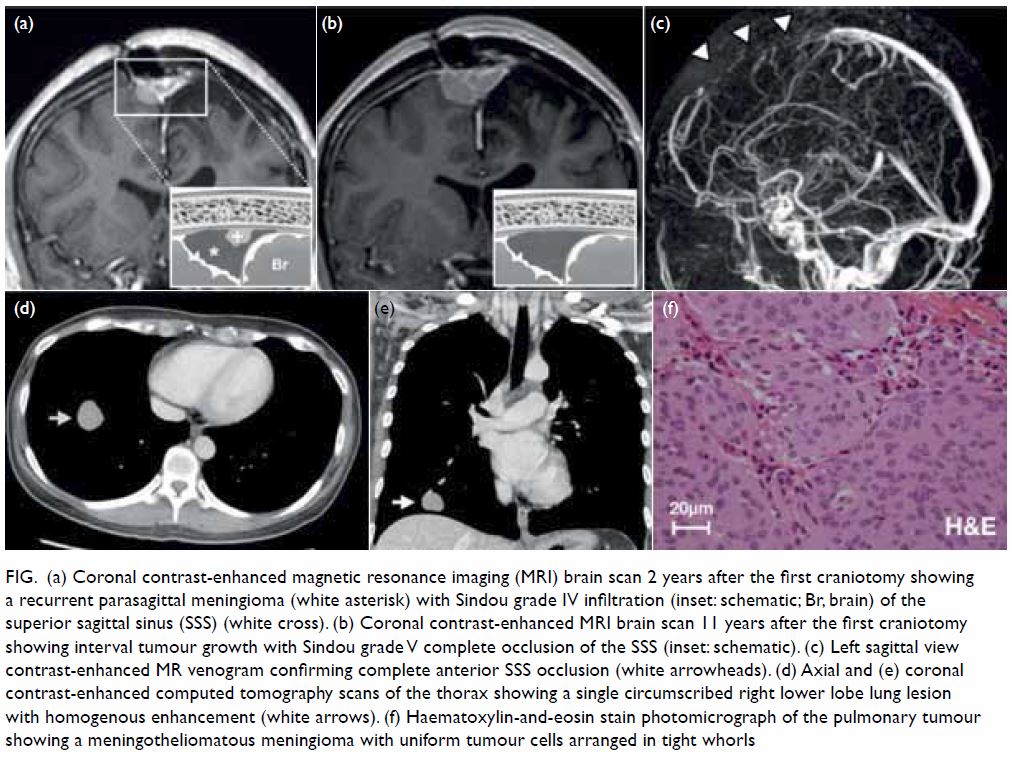
at the right hypoglossal canal (Fig 1).
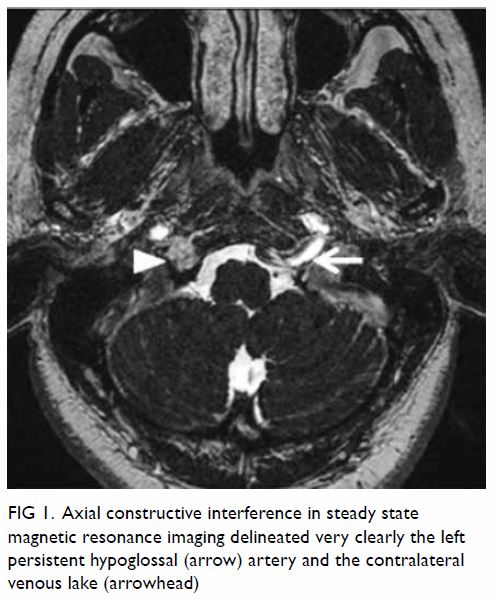
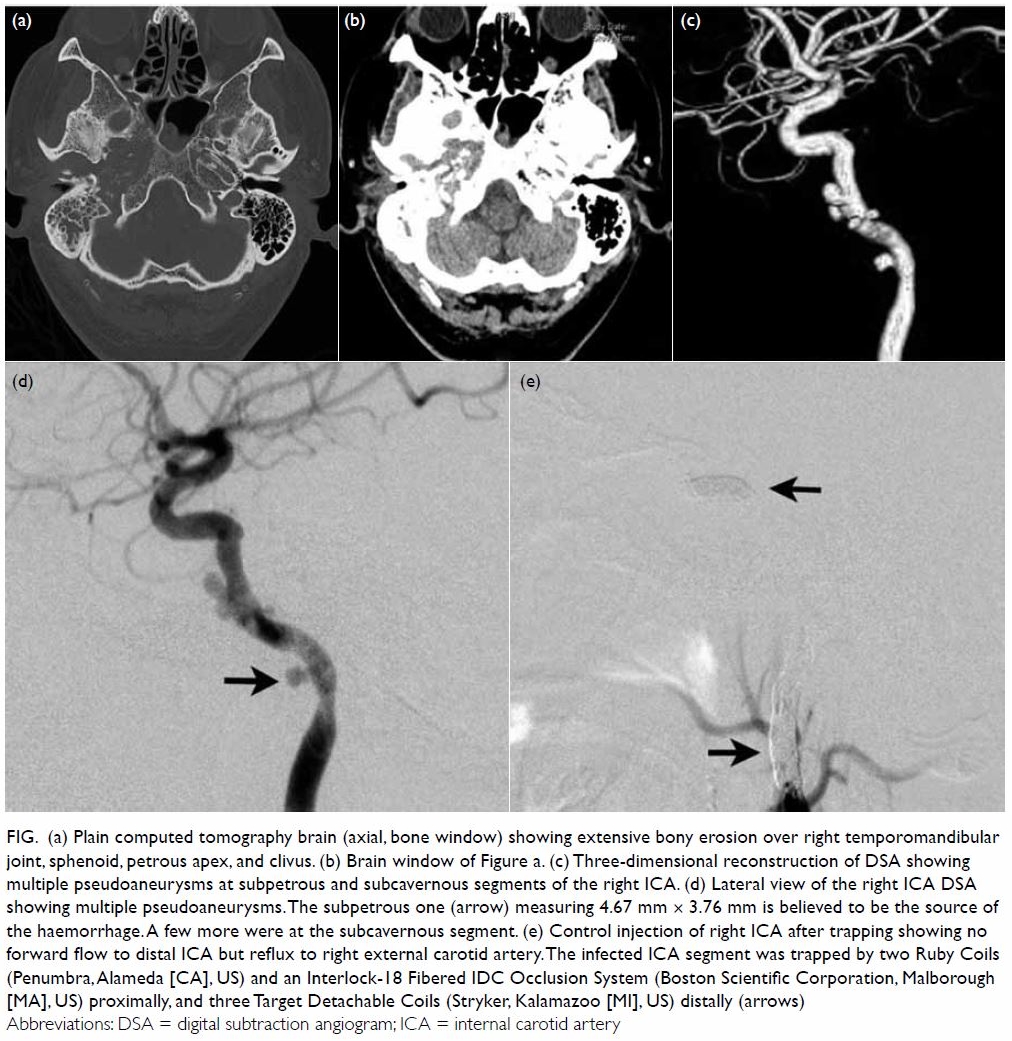
Figure 1. Axial constructive interference in steady state magnetic resonance imaging delineated very clearly the left persistent hypoglossal (arrow) artery and the contralateral venous lake (arrowhead)
Additionally, an anomalous vessel arising from the
left internal carotid artery at C2 level was noted, entering the cranium
through the left hypoglossal canal and joining the basilar artery. This
anomalous vessel corresponded to a left persistent hypoglossal artery
(PHA; Figs 1 and 2). The bilateral cervical vertebral arteries were
diminutive in calibre and did not serve as major arterial supplies to the
basilar artery. No intracranial aneurysms were detected and no infarction
or other abnormality was noted. The patient’s symptoms later substantially
improved with symptomatic treatment.
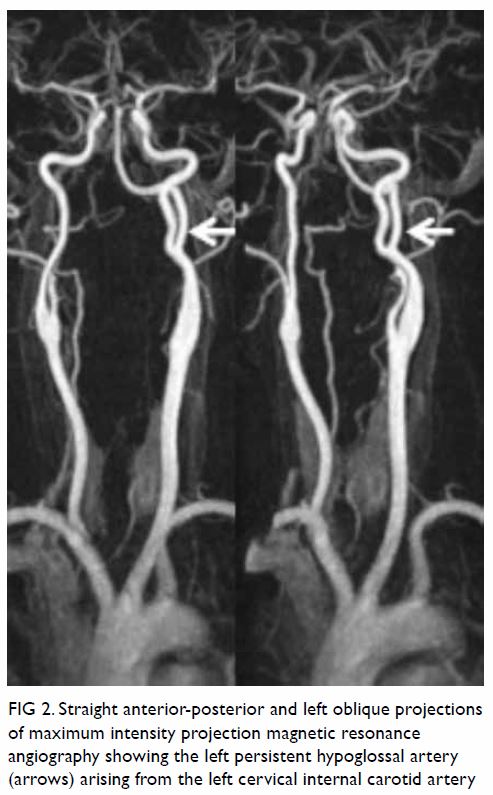
Figure 2. Straight anterior-posterior and left oblique projections of maximum intensity projection magnetic resonance angiography showing the left persistent hypoglossal artery (arrows) arising from the left cervical internal carotid artery
Discussion
Bony venous lakes of the skull are common and
asymptomatic, and they are typically parasagittal in location. In CT
scans, they appear as lucent lesions with corticated/sclerotic margins. In
MR imaging, they exhibit the same signal characteristics as veins.
However, it is rare to find venous lakes located at the hypoglossal canal,
and other entities must be excluded such as a neurinoma or even a dural
arteriovenous fistula of the hypoglossal canal, another rare but
potentially symptomatic condition that may follow head trauma.1 In this case, there was no apparent arteriovenous shunt
detected in the MR angiography sequences.
A PHA results from failure of regression of a
primitive hypoglossal artery, one of the several anastomoses that exist
between the carotid and vertebrobasilar arteries during embryogenesis.
Although rare, it is the second most common persistent
carotid-vertebrobasilar anastomosis after the trigeminal artery, with a
prevalence of up to 0.29%,2 usually
representing an incidental finding. However, diagnosis of PHA is important
because it is often the only blood supply to the basilar trunk, as
vertebral arteries are usually hypoplastic. Moreover, PHA is associated
with intracranial arterial aneurysms, ischaemic cerebrovascular attacks,
subarachnoid haemorrhage and arteriovenous malformations.3 Recognition of PHA is extremely important before any
endovascular procedure, carotid endarterectomy or skull base surgery is
performed. Exposure of the basilar trunk to an unusual haemodynamic stress
could be the underlying mechanism that predisposes an individual to the
development of aneurysms.4 On the
contrary, there is an increased risk of ischaemia caused by embolism from
the internal carotid artery to the posterior circulation through the PHA.5
Both vascular anomalies in this patient were most
likely incidental findings; however, owing to the reported association of
PHA with intracranial aneurysm development and ischaemic events, any new
episode or the development of neurological symptoms should have triggered
immediate imaging study.
To the best of our knowledge, this is the first
report of a PHA with a contralateral hypoglossal canal venous lake, both
representing rare vascular variants.
Author contributions
All authors contributed to the concept of study,
acquisition and analysis of data, drafting of the article, and critical
revision for important intellectual content. All authors had full access
to the data, contributed to the study, approved the final version for
publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of
interest.
Funding/support
This case report received no specific grant from
any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the
Declaration of Helsinki. The patient provided informed consent for all
procedures.
References
1. Manabe S, Satoh K, Matsubara S, Satomi
J, Hanaoka M, Nagahiro S. Characteristics, diagnosis and treatment of
hypoglossal canal dural arteriovenous fistula: report of nine cases.
Neuroradiology 2008;50:715-21. Crossref
2. Uchino A, Saito N, Okada Y, et al.
Persistent hypoglossal artery and its variants diagnosed by CT and MR
angiography. Neuroradiology 2013;55:17-23. Crossref
3. Srinivas MR, Vedaraju KS, Manjappa BH,
Nagaraj BR. Persistent primitive hypoglossal artery (PPHA)—a rare anomaly
with literature review. J Clin Diagn Res 2016;10:TD13-4.
4. Terayama R, Toyokuni Y, Nakagawa S, et
al. Persistent hypoglossal artery with hypoplasia of the vertebral and
posterior communicating arteries. Anat Sci Int 2011;86:58-61. Crossref
5. Conforto AB, de Souza M, Puglia P Jr,
Yamamoto FI, da Costa Leite C, Scaff M. Bilateral occipital infarcts
associated with carotid atherosclerosis and a persistent hypoglossal
artery. Clin Neurol Neurosurg 2007;109:364-7. Crossref