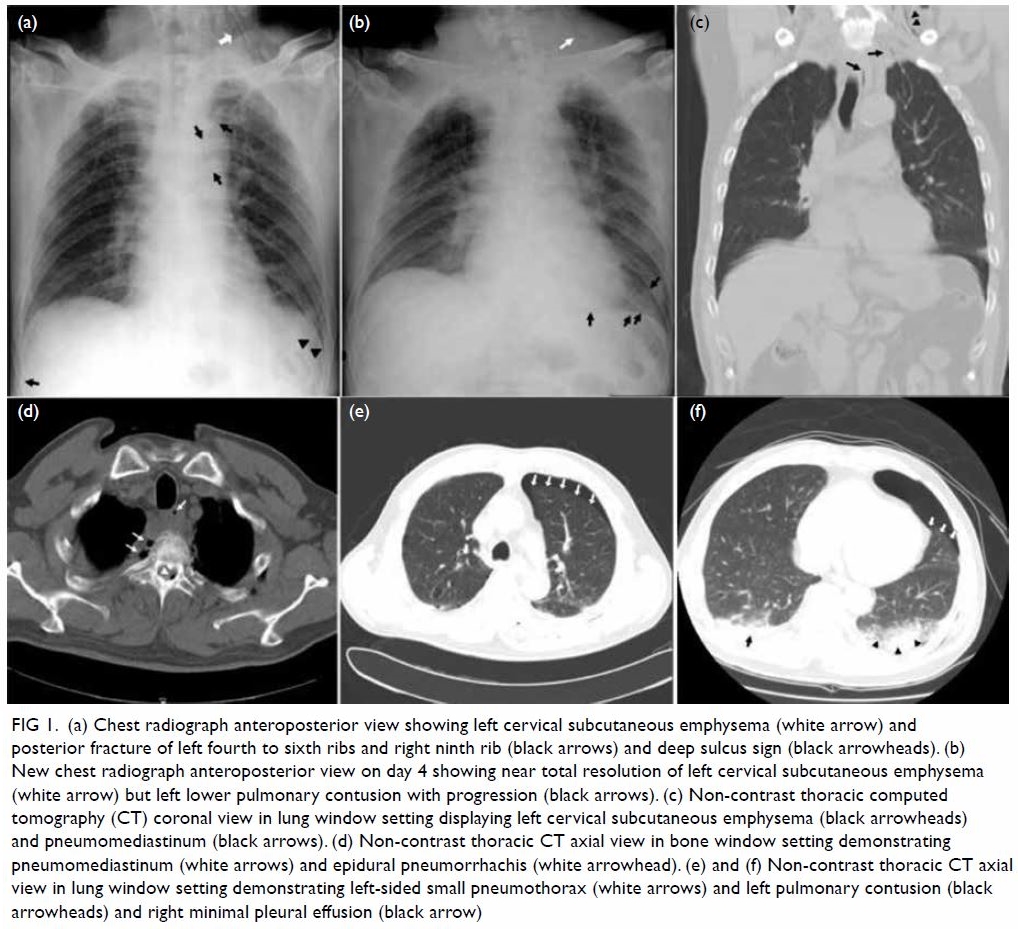
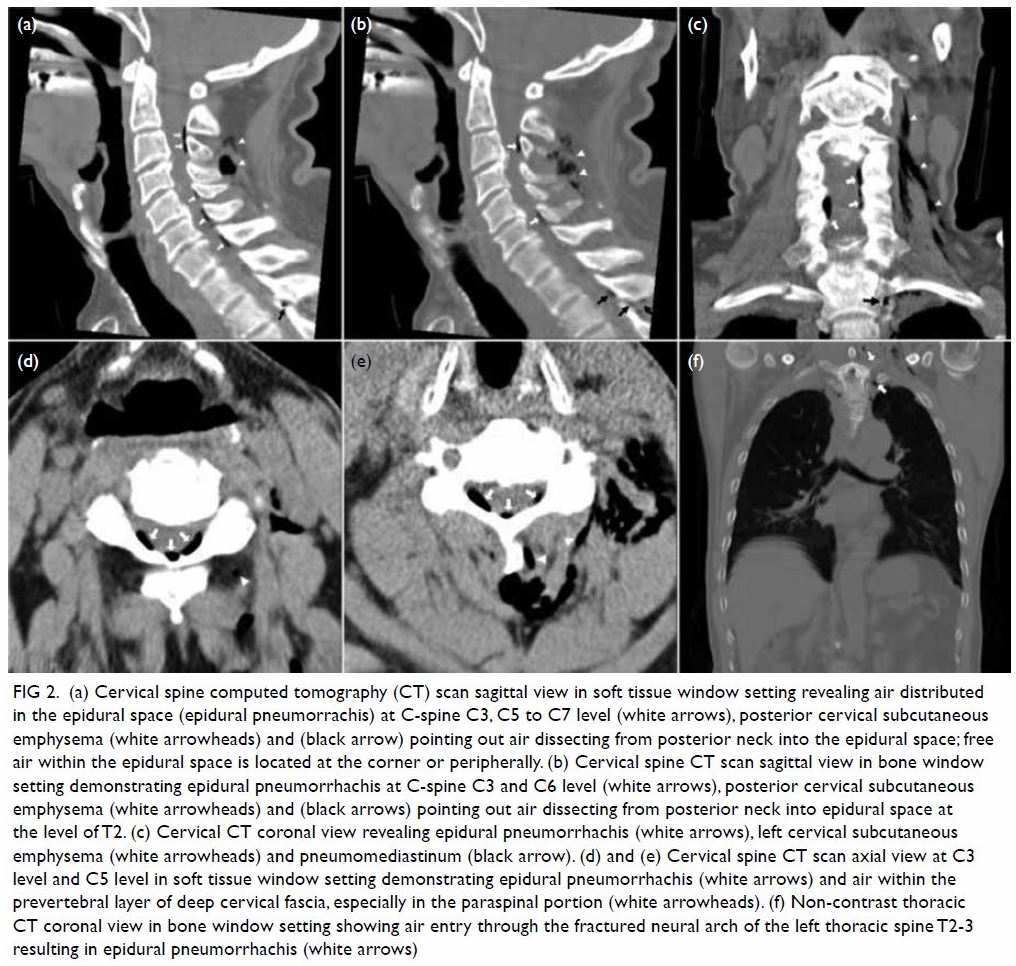
Herpes zoster radiculopathy as a rare cause of foot drop: a case report
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Herpes zoster radiculopathy as a rare cause of
foot drop: a case report
Gene Leung, MB, ChB, MRCSEd
Department of Orthopaedics and Traumatology, Pamela Youde Nethersole Eastern Hospital, Hong Kong
Corresponding author: Dr Gene Leung (geneleung@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Case report
A 69-year-old woman with a medical history of
hypertension and hyperlipidaemia presented to
the Medicine and Geriatrics unit of Pamela Youde
Nethersole Eastern Hospital with acute onset of
right leg rash for 5 days. The rash was associated with
burning pain and dysesthesia along the right leg.
There was no back pain and no history of back injury.
She was initially diagnosed with a herpes zoster
infection and treated with a course of acyclovir and
amitriptyline and then discharged home. The patient
presented again 10 days later for new-onset right big
toe weakness with right foot drop. On the second
admission, an orthopaedic consultation was sought.
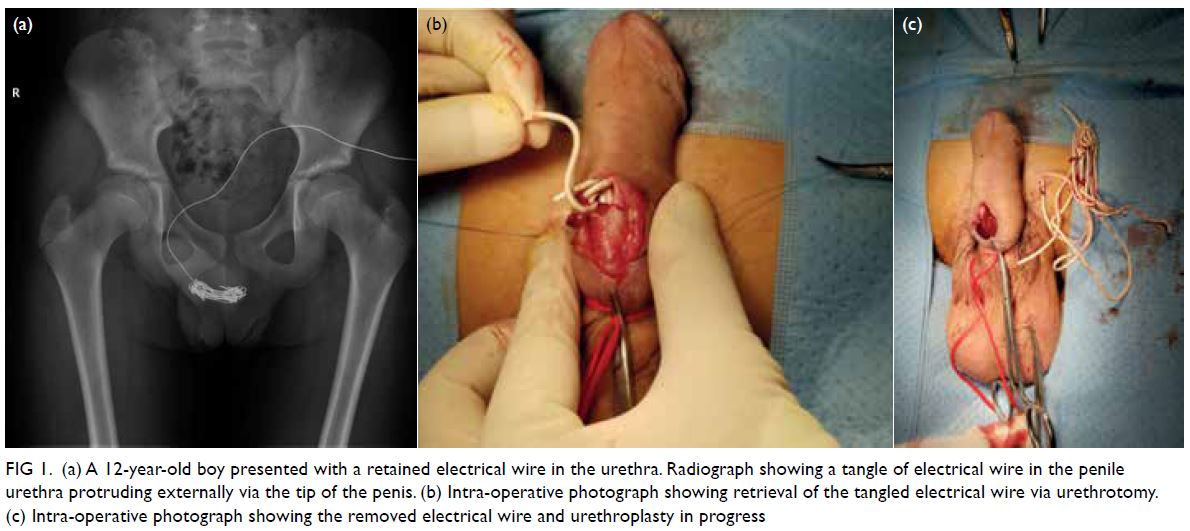
Physical examination revealed a cutaneous
papulovesicular rash and vesicles along the lateral
aspect of her right leg down to the dorsum of her
right foot (Fig 1). The rash corresponded to an L5
dermatomal distribution. Testing of sensation
showed hyperesthesia along the same region.
Right hip abduction (gluteus medius) power was
Medical Research Council (MRC) grade 4. Right
hip flexion (iliopsoas), right hip extension (gluteus
maximus), right knee flexion (hamstrings), right
knee extension (quadriceps) and right ankle plantar
flexion (gastrocnemius) demonstrated full power
of MRC grade 5. She had right foot drop with right
ankle dorsiflexion (tibialis anterior tendon), MRC
grade 2. Right big toe dorsiflexion (extensor hallucis
longus) showed a power of MRC grade 1. Right big
toe plantar flexion (flexor hallucis longus) tested
MRC grade 4 power. Right ankle inversion (tibialis
posterior) and right ankle eversion (peronei) elicited
MRC grade 3 power. Lasègue’s sign was positive for
the right lower limb but there was no tenderness
along the spine. There were no neurological deficits
of her left lower limb. Achilles tendon and patellar
tendon reflexes were normal, and sphincter function
was not disturbed.
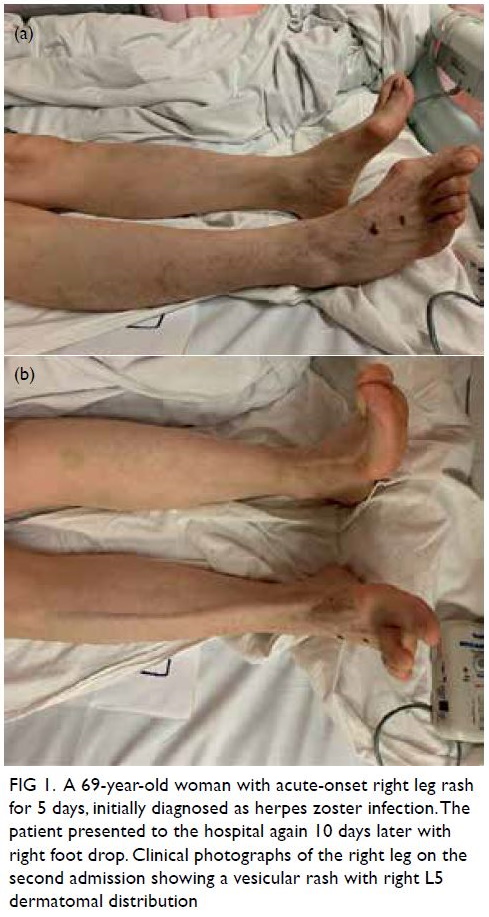
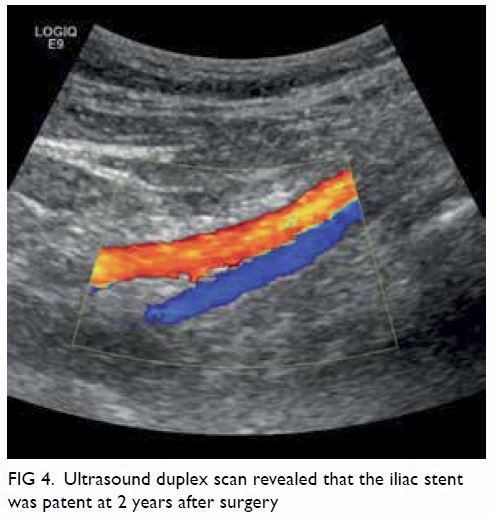
Figure 1. A 69-year-old woman with acute-onset right leg rash for 5 days, initially diagnosed as herpes zoster infection. The patient presented to the hospital again 10 days later with right foot drop. Clinical photographs of the right leg on the second admission showing a vesicular rash with right L5 dermatomal distribution
Plain radiographs of the lumbosacral spine
showed mild degeneration over the lower lumbar
facet joints. The pedicles and endplates were intact
with no vertebral collapse or slippage. The patient
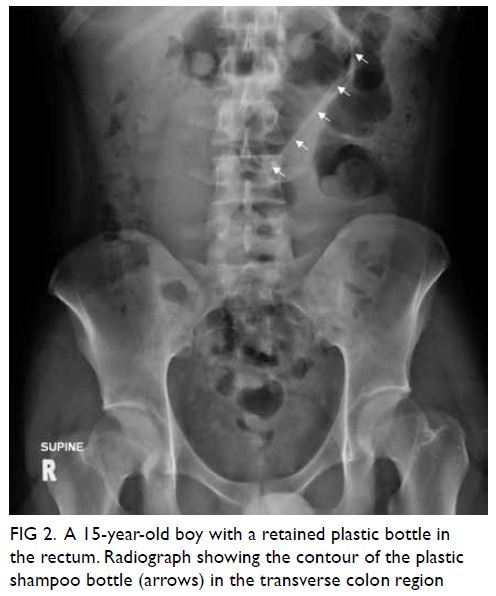
was referred for magnetic resonance imaging of
the lumbosacral spine to exclude concomitant
compression of the neurological elements (Fig 2). There was no intervertebral disc prolapse or nerve root impingement and lateral recess and
intervertebral foramen were not stenotic.
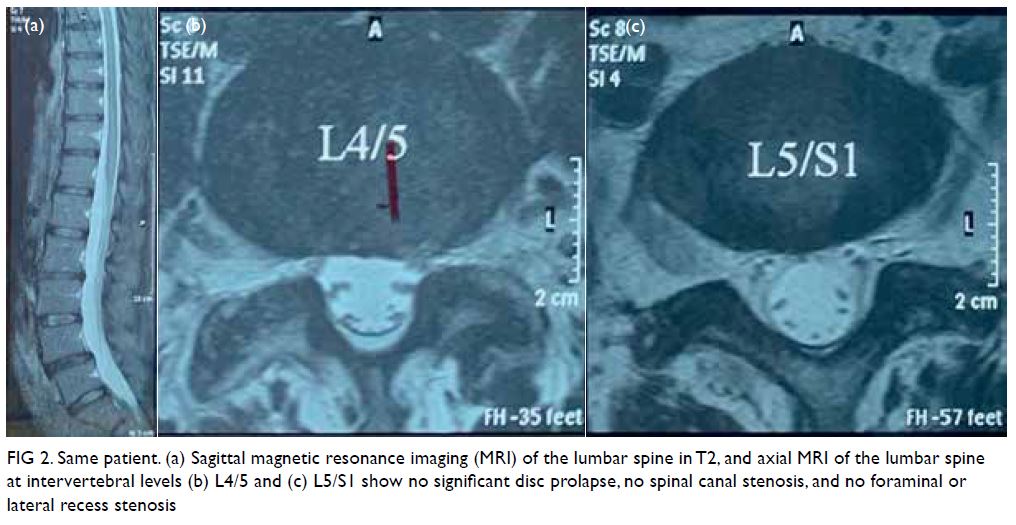
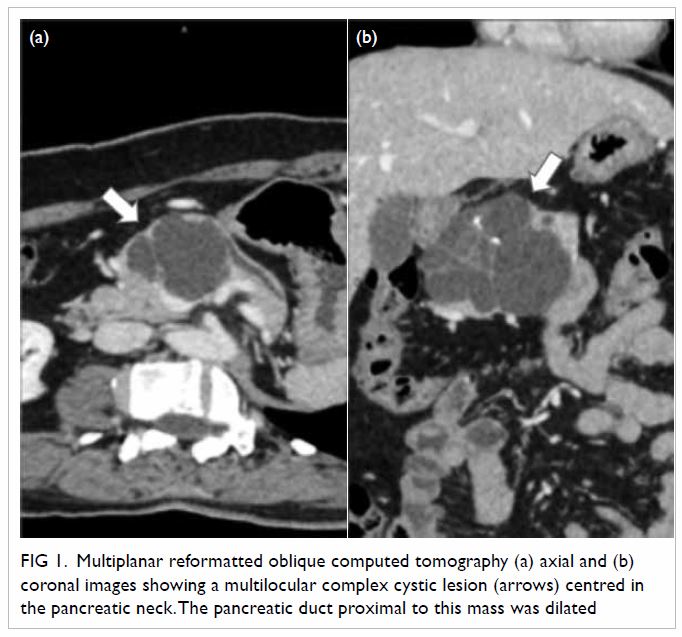
Figure 2. Same patient. (a) Sagittal magnetic resonance imaging (MRI) of the lumbar spine in T2, and axial MRI of the lumbar spine at intervertebral levels (b) L4/5 and (c) L5/S1 show no significant disc prolapse, no spinal canal stenosis, and no foraminal or lateral recess stenosis
Pain medication was stepped up to gabapentin.
She was discharged with a course of out-patient
physiotherapy.
At 6-week follow-up examination, the vesicles
had crusted, and the neuropathic pain had subsided.
The power of right sided hip abduction, big toe plantar flexion, ankle plantarflexion, ankle inversion
and ankle eversion now returned to full. There was
residual weakness in right ankle dorsiflexion, with a
power of MRC grade 4, and right big toe dorsiflexion,
with a power of MRC grade 2.
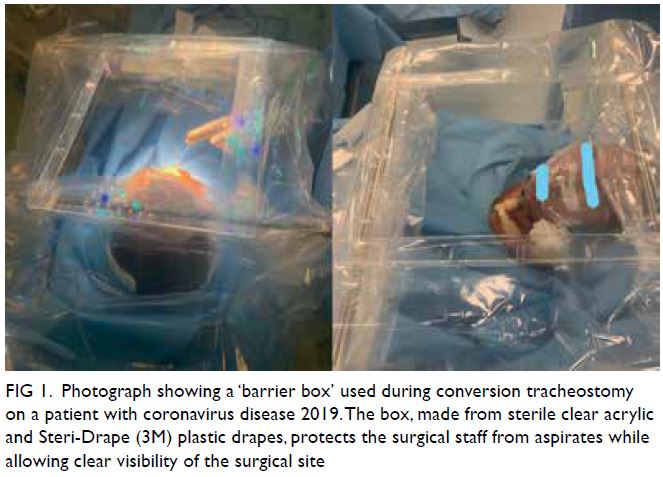
At 3-month follow-up examination, the rashes
over the right leg and foot had disappeared, leaving
faint pigmentation (Fig 3). She was pain free. Right
ankle dorsiflexion power was full and right big toe
dorsiflexion power further improved to MRC grade 3.
She was weaned off gabapentin and continued with
physiotherapy for ankle strengthening exercises.
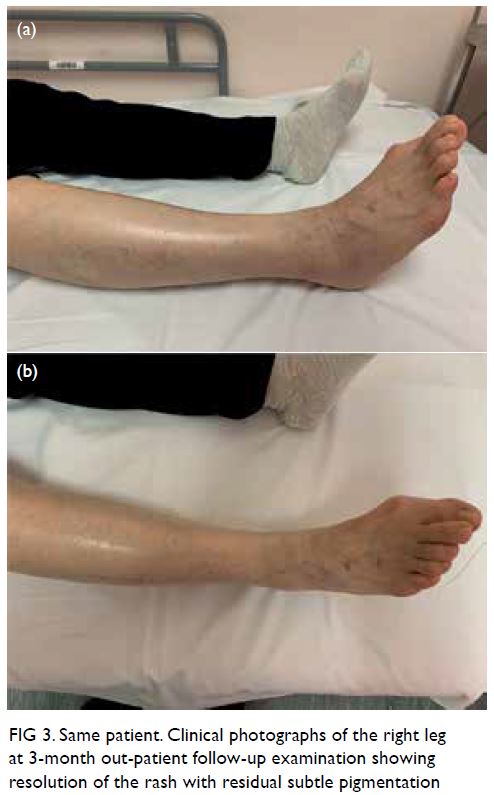
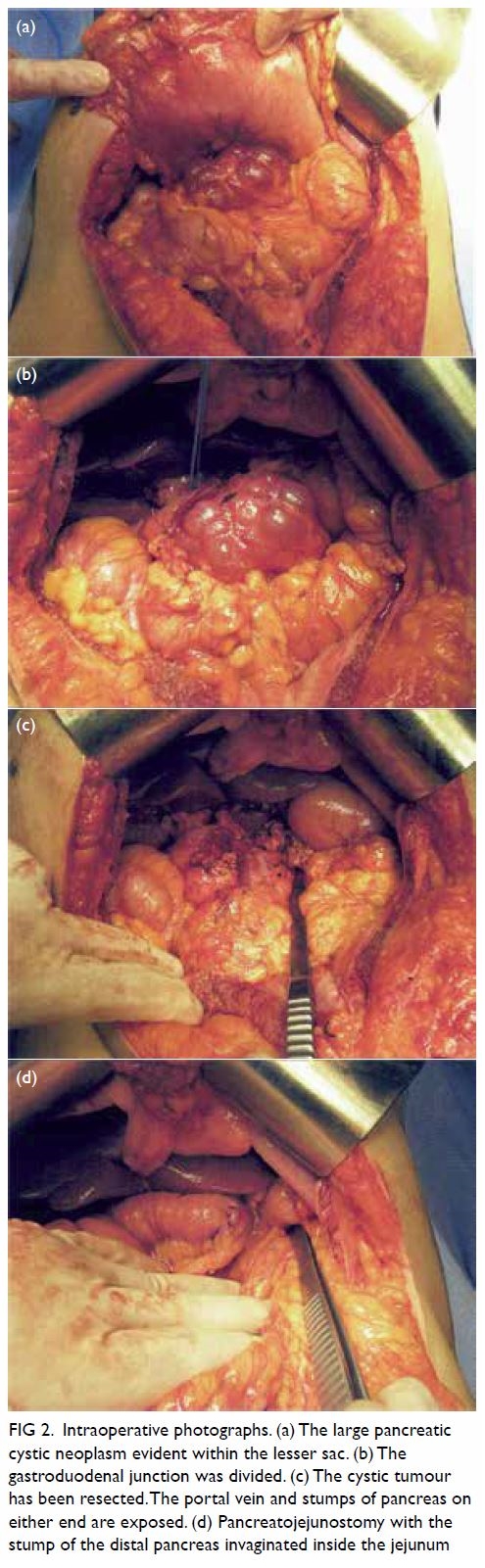
Figure 3. Same patient. Clinical photographs of the right leg at 3-month out-patient follow-up examination showing resolution of the rash with residual subtle pigmentation
Discussion
Herpes zoster is a viral infection of the nervous
system that results from the reactivation of a
previous varicella zoster virus infection, often in
childhood. The virus lays dormant in the dorsal
root ganglion re-emerging as the patient ages or
becomes immunocompromised.1 It manifests with
a typical cutaneous vesicular rash that follows a
spinal nerve distribution. Classically, it is believed
that the virus spreads from the dorsal root ganglion
in an antegrade fashion along the spinal nerve.
Thus, symptoms are most often purely sensory.
Rarely, when motor manifestations are present, they
tend to affect more proximal muscle groups.2 The
typical example is Bell’s palsy due to facial nerve
involvement. It has been hypothesised that local
inflammation of the dorsal root ganglion results in
hypervascularity in the surrounding nerve tissue,
disrupting the blood-nerve barrier which can also
result in a motor deficit.3 Therefore, although post-herpetic
neuralgia is a common manifestation of
herpes zoster infection, segmental zoster paresis is rare. Although reported incidences in different
countries vary, a recent study performed with a
majority of Chinese patients reported paresis in only 0.57% of those afflicted with a zoster infection.2 Since
lumbar radiculopathy resulting from intervertebral
disc herniation with nerve root irrigation commonly
involves L4/5 and L5/S1 levels, a herpes zoster
infection with segmental paresis affecting the L5
spinal nerve is a close mimicker. In some instances,
the motor weakness can precede the rash.4
The diagnosis of herpes zoster radiculopathy is
clinical although it can be aided by serum antibodies,
electrodiagnostic studies and formal dermatological
assessment. For this case, although electrodiagnostic
testing would have been ideal to supplement the
overall assessment, it was not arranged due to the
anticipated recovery and resource limitations in
the public hospital setting. As the lumbar spinal
nerve roots were involved, the clinical presentation
resembled compressive pathologies that are
commonly seen in orthopaedics. A plain magnetic
resonance imaging can exclude the presence of
nerve root impingement, although findings that may
suggest zoster infection such as spinal nerve swelling
with T2 hyperintensity are neither sensitive nor
specific.3
The mainstay of treatment for zoster
radiculopathy is pharmacological. A course of
antivirals is prescribed for the acute herpetic
infection. Pain is managed according to the World
Health Organization analgesic ladder or with specific
neuropathic pain medication such as amitriptyline,
gabapentin or pregabalin. Although the clinical
course is variable, most patients can expect to make
a near-complete motor recovery within 1 year.2 A
well-fitted ankle-foot orthosis facilitates ambulation
if there is persistent foot drop. In addition,
transforaminal epidural steroid injections have been
reported to mitigate pain and weakness by suppressing
inflammation along the spinal root.5 However, the use of corticosteroids remains controversial as there
is a theoretical risk of viral reactivation.3
Author contributions
The author contributed to the concept of the study, acquisition
and analysis of data, drafting of the manuscript, and critical
revision for important intellectual content. The author had
full access to the data, contributed to the study, approved the
final version for publication, and takes responsibility for its
accuracy and integrity.
Conflicts of interest
The author has disclosed no conflicts of interest.
Funding/support
This case report received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of Helsinki. The patient provided informed consent for all
procedures.
References
1. Hope-Simpson RE. The nature of herpes zoster: a long-term study and a new hypothesis. Proc R Soc Med 1965;58:9-20. Crossref
2. Liu Y, Wu BY, Ma ZS, et al. A retrospective case series of segmental zoster paresis of limbs: clinical,
electrophysiological and imaging characteristics. BMC
Neurol 2018;18:121. Crossref
3. Hackenberg RK, von den Driesch A, König DP. Lower back
pain with sciatic disorder following L5 dermatome caused
by herpes zoster infection. Orthop Rev (Pavia) 2015;7:6046. Crossref
4. Teo HK, Chawla M, Kaushik M. A rare complication of herpes zoster: segmental zoster paresis. Case Rep Med.
2016;2016:7827140. Crossref
5. Conliffe TD, Dholakia M, Broyer Z. Herpes zoster
radiculopathy treated with fluoroscopically-guided selective
nerve root injection. Pain Physician 2009;12:851-3. Crossref