Hong Kong Med J 2015 Dec;21(6):573.e3–5
DOI: 10.12809/hkmj154728
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
PICTORIAL MEDICINE
The nail points to the diagnosis
Stephanie YK Tong, BSc;
HM Luk, FHKAM (Paediatrics);
Tony MF Tong, MSc;
Ivan FM Lo, FHKAM (Paediatrics)
Clinical Genetic Service, Department of Health, Hong Kong
Corresponding author: Dr HM Luk (luksite@gmail.com)
 Full
paper in PDF
Full
paper in PDF
A 54-year-old man was referred to the genetic clinic
with familial nail dysplasia in September 2012 (Fig 1).
On further questioning, he reported recurrent knee
pain due to patellar dislocation. There was no elbow
involvement or renal problem. A skeletal survey
was performed in view of his skeletal complaint (Fig 2). His family history was significant: his paternal
grandfather, father, and two of his paternal uncles
also had nail dysplasia and knee problems, but had
undergone no formal medical assessment. What is
the diagnosis?
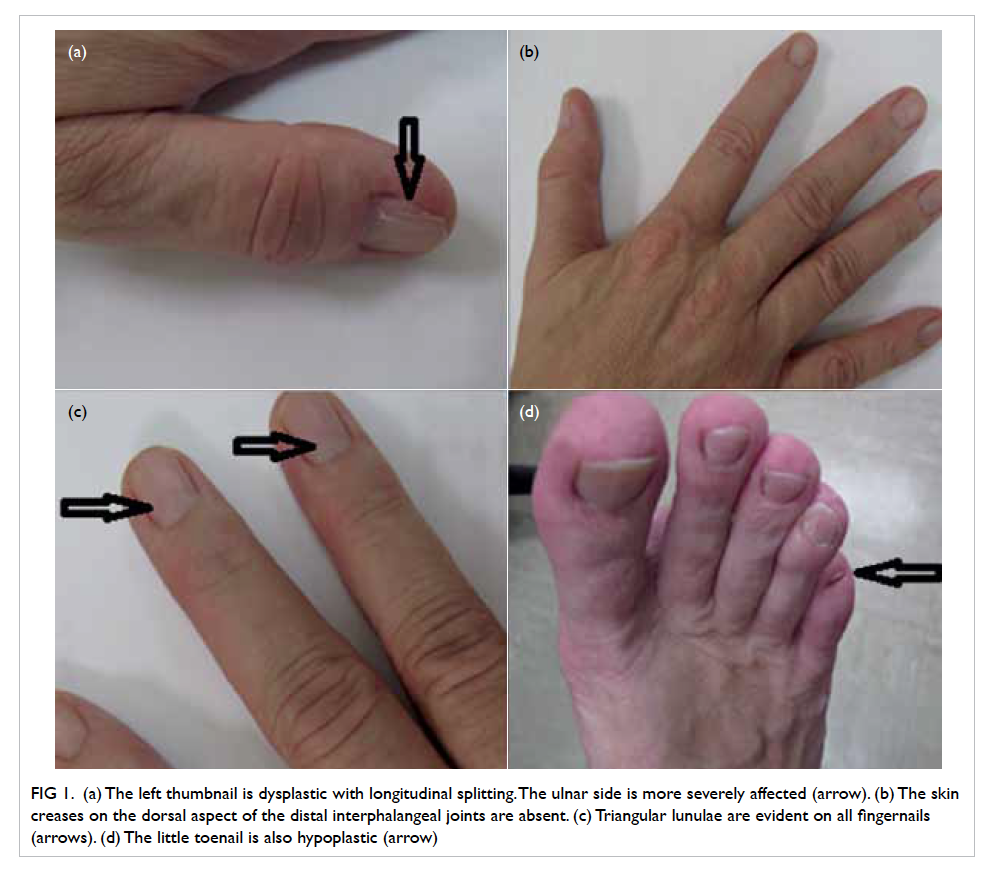
Figure 1. (a) The left thumbnail is dysplastic with longitudinal splitting. The ulnar side is more severely affected (arrow). (b) The skin creases on the dorsal aspect of the distal interphalangeal joints are absent. (c) Triangular lunulae are evident on all fingernails (arrows). (d) The little toenail is also hypoplastic (arrow)
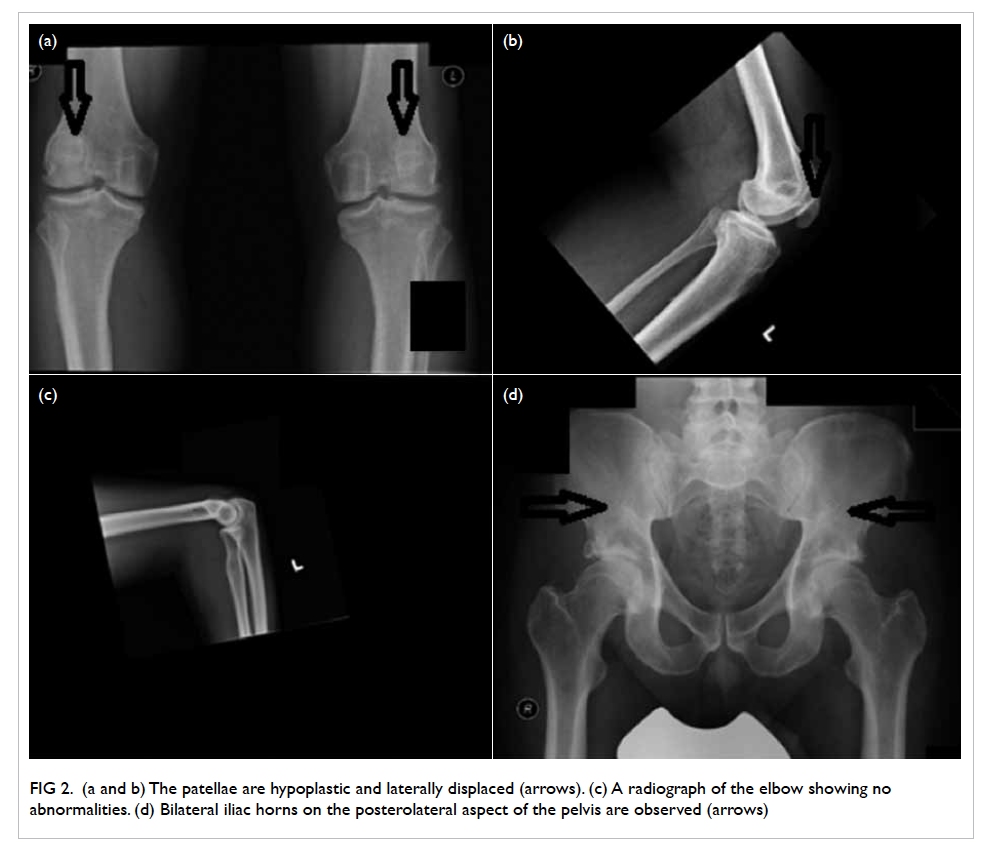
Figure 2. (a and b) The patellae are hypoplastic and laterally displaced (arrows). (c) A radiograph of the elbow showing no abnormalities. (d) Bilateral iliac horns on the posterolateral aspect of the pelvis are observed (arrows)
What is his diagnosis?
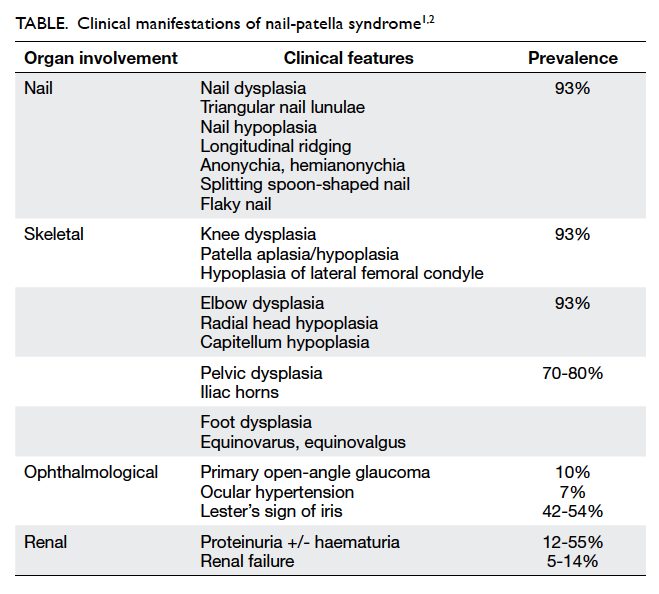
The combination of nail dysplasia, patella hypoplasia,
and iliac horn led to the clinical diagnosis of
nail-patella syndrome (NPS). This syndrome is
also known as Fong disease or hereditary osteo-onychodysplasia.
Multiple organ systems are
affected including the nails, the eyes, the kidneys,
and the skeleton. The clinical presentation of NPS
can be highly heterogeneous and is summarised in
the Table.1 2 It is a rare autosomal dominant disease
with a prevalence of about 1 in 50 000 newborn.1 It is
highly penetrant but with significant intra- and inter-familial
variability in expression.
Is there any genetic testing available for such condition and what is the underlying pathogenesis?
The diagnosis of NPS is usually based on clinical
findings. It is straightforward when the classic tetrad
of abnormal nails, elbows, knees, and iliac horns
are present. Nonetheless molecular genetic testing
should be considered when the diagnosis is in doubt,
or when prenatal or pre-implantation diagnosis is
desired. The LMX1B gene is the only gene known
to be associated with NPS. Sequence analysis would
identify the LMX1B gene mutation in 85% of cases
of NPS.
LMX1B gene sequencing was performed for
this patient (Fig 3) and revealed a missense mutation
LMX1B NM_002316.3}:c.[175T>C];[=];LMX1B{NP
_002307.2}:p.[Cys59Arg];[=]. This changed the 59th
amino acid from cysteine to arginine in the LIM-A
domain of the LMX1B protein. It was located in an
evolutionarily highly conserved region and has been
reported in the literature to be associated with NPS.
Therefore, it was considered to be pathogenic.3
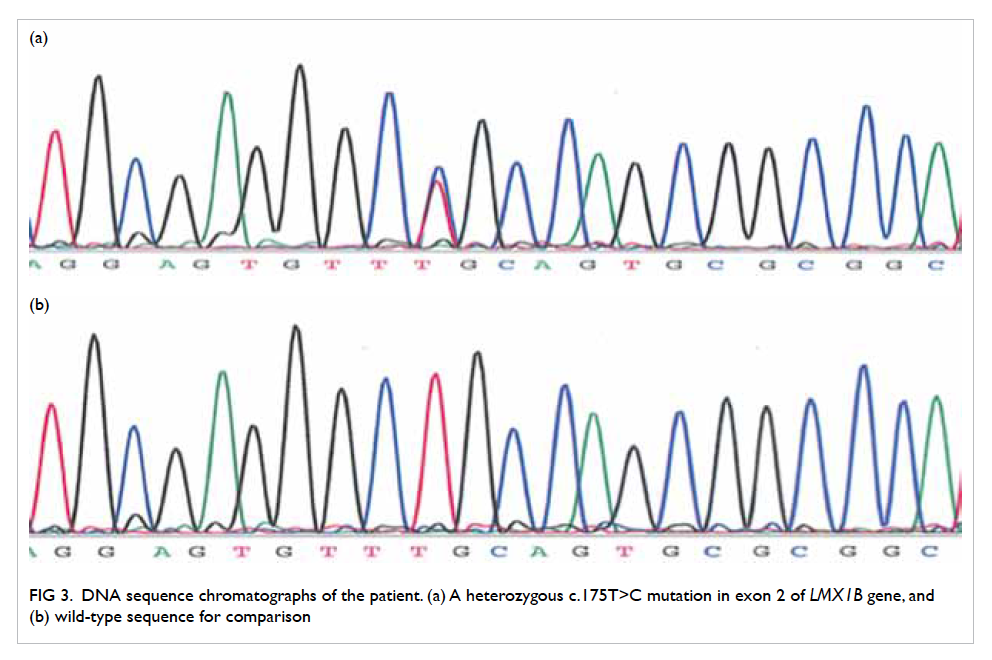
Figure 3. DNA sequence chromatographs of the patient. (a) A heterozygous c.175T>C mutation in exon 2 of LMX1B gene, and (b) wild-type sequence for comparison
To date, more than 150 mutations of the LMX1B
gene have been reported, but no clear genotype-phenotype
correlation has been demonstrated.
LMX1B has been demonstrated in animal models
to be involved in multiple developmental functions
including dorso-ventral patterning of the limb
bud, and cellular differentiation in the kidney.
Nonetheless the exact pathogenesis of NPS has not
been elucidated.4
How to manage this patient?
Upon initial diagnosis, comprehensive renal,
orthopaedic, and ophthalmological assessments are
essential. The renal manifestation strongly affects the
long-term prognosis. Kidney involvement occurs in
30% to 50% of patients, but kidney failure only occurs
in 3% to 5%.2 Since nephropathy may not develop
until later in life, prospective monitoring is essential
for all NPS patients. Primary open-angle glaucoma or
ocular hypertension occurs in 10% of NPS patients
so regular eye surveillance is also warranted. Nail-patella
syndrome is an autosomal dominant disorder
with a 50% chance of occurrence in offspring, thus
genetic counselling is essential (Box2). Genetic testing of other at-risk family members, and prenatal and
pre-implantation genetic diagnoses are possible only
if the disease-causing mutation is known in the index
patient.

References
1. Bongers EM, Gubler MC, Knoers NV. Nail-patella
syndrome. Overview on clinical and molecular findings.
Pediatr Nephrol 2002;17:703-12. Crossref
2. Sweeney E, Fryer A, Mountford R, Green A, McIntosh I.
Nail-patella syndrome: a review of the phenotype aided by
developmental biology. J Med Genet 2003;40:153-62. Crossref
3. Clough MV, Hamlington JD, McIntosh I. Restricted
distribution of loss-of-function mutations within the
LMX1B genes of nail-patella syndrome patients. Hum
Mutat 1999;14:459-65. Crossref
4. Chen H, Lun Y, Ovchinnikov D, et al. Limb and kidney
defects in LMX1B mutant mice suggest an involvement
of LMX1B in human nail patella syndrome. Nat Genet
1998;19:51-5. Crossref