© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Infantile to late adulthood onset facioscapulohumeral dystrophy type 1: a case series
WY Leung, MB, BS, MRCPCH1; HM Luk, FHKCPaed, FHKAM (Paediatrics)2; Varut Vardhanabhuti, PhD (UK), FRCR (UK)3; Y Gao, PhD, FHKCP (Neurology), FHKAM (Medicine)4; KF Hui, FHKCP (Neurology), FHKAM (Medicine)5; WY Lau, FHKCP (Neurology), FHKAM (Medicine)6; Terence PH Young, FHKCP (Neurology), FHKAM (Medicine)7; Jessica TC Li, FHKCP (Neurology), FHKAM (Medicine)8; Eva LW Fung, FHKCPaed, FHKAM (Paediatrics)9; Annie TG Chiu, FHKCPaed, FHKAM (Paediatrics)1; Ivan FM Lo, FHKCPaed, FHKAM (Paediatrics)2; Brian HY Chung, FHKAM (Paediatrics), FCCMG (Clinical Genetics, Canada)1; YF Cheung, FHKCP (Neurology), FHKAM (Medicine)8; Sophelia HS Chan, FHKCPaed, FHKAM (Paediatrics)1
1 Department of Paediatrics and Adolescent Medicine, Queen Mary Hospital, The University of Hong Kong, Hong Kong
2 Clinical Genetic Service, Department of Health, Hong Kong SAR Government, Hong Kong
3 Department of Diagnostic Radiology, The University of Hong Kong, Hong Kong
4 Department of Medicine, Queen Mary Hospital, Hong Kong
5 Department of Medicine and Geriatrics, United Christian Hospital, Hong Kong
6 Department of Medicine and Geriatrics, Kwong Wah Hospital, Hong Kong
7 Department of Medicine and Geriatrics, Ruttonjee & Tang Shiu Kin Hospitals, Hong Kong
8 Department of Medicine, Queen Elizabeth Hospital, Hong Kong
9 Department of Paediatrics, Prince of Wales Hospital, Hong Kong
Corresponding author: Dr Sophelia HS Chan (sophehs@hku.hk)
 Full paper in PDF
Full paper in PDF
Cases
We present the clinical data of eight patients
with genetically confirmed facioscapulohumeral
muscular dystrophy type 1 (FSHD1) in Hong Kong
(Table).
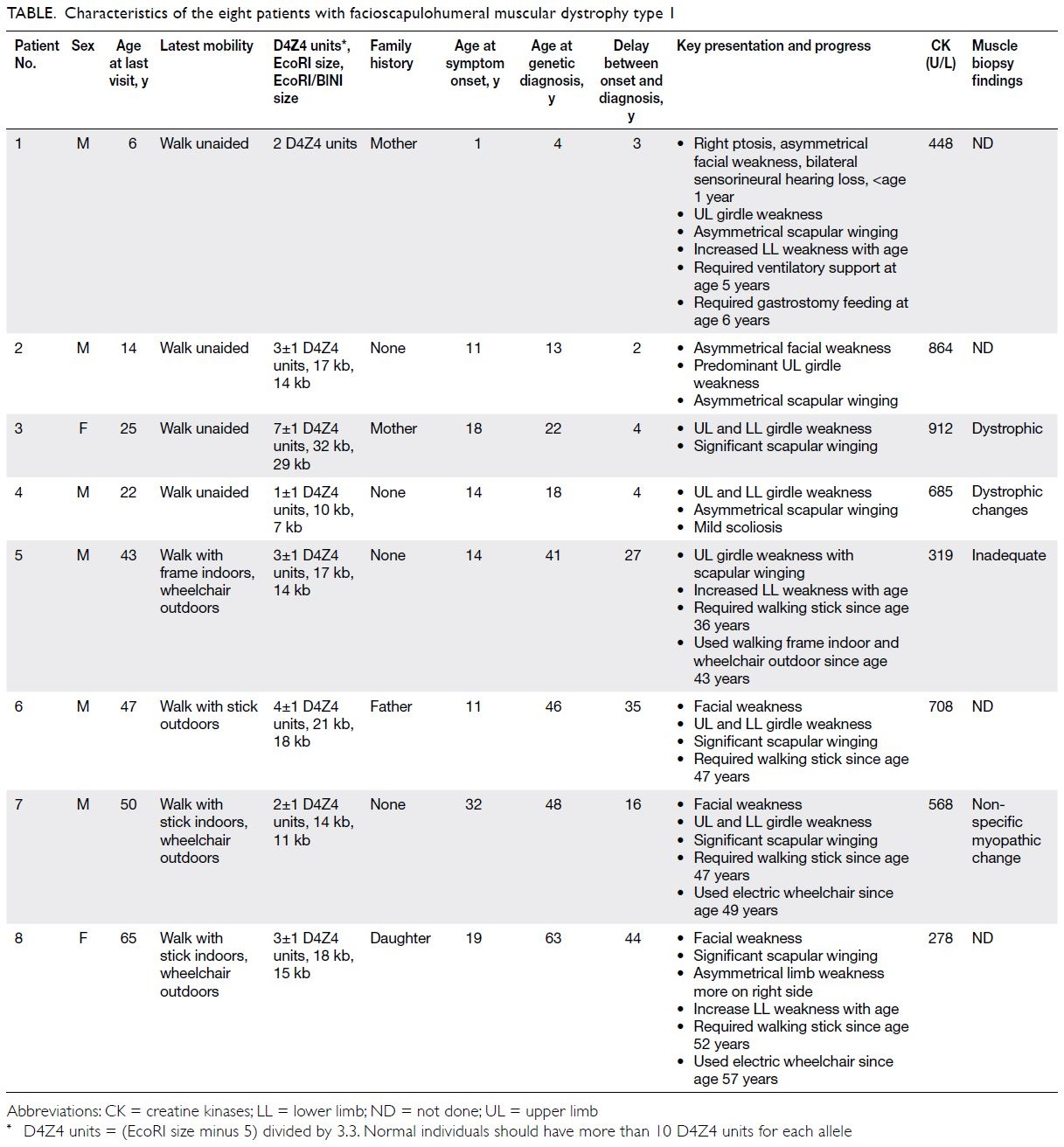
Table. Characteristics of the eight patients with facioscapulohumeral muscular dystrophy type 1
In June 2015, Patient 1 presented with right
ptosis and bilateral sensorineural hearing loss at age
1 year. He had early-onset FSHD1 of the most severe
phenotype, with early development of asymmetrical
facial and upper limb weakness, significant hearing
impairment requiring hearing aids, and an early
need for ventilation and feeding support (Fig 1).
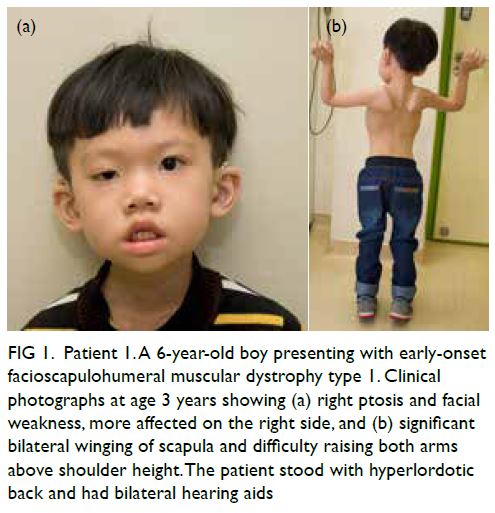
Figure 1. Patient 1. A 6-year-old boy presenting with early-onset facioscapulohumeral muscular dystrophy type 1. Clinical photographs at age 3 years showing (a) right ptosis and facial weakness, more affected on the right side, and (b) significant bilateral winging of scapula and difficulty raising both arms above shoulder height. The patient stood with hyperlordotic back and had bilateral hearing aids
In May 2019, Patient 2 presented with
progressive asymmetrical facial weakness with
predominant upper limb girdle weakness from age
11 years (Fig 2). Magnetic resonance imaging scan
was taken at age 13 years (Fig 3).
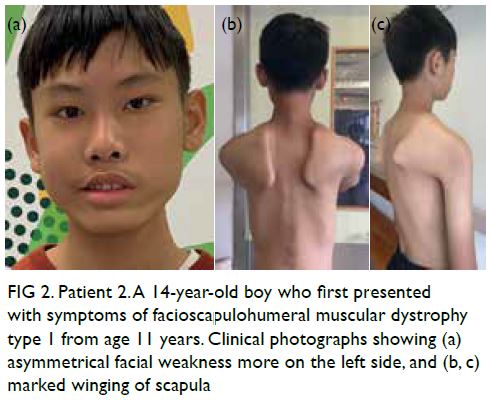
Figure 2. Patient 2. A 14-year-old boy who first presented with symptoms of facioscapulohumeral muscular dystrophy type 1 from age 11 years. Clinical photographs showing (a) asymmetrical facial weakness more on the left side, and (b, c) marked winging of scapula
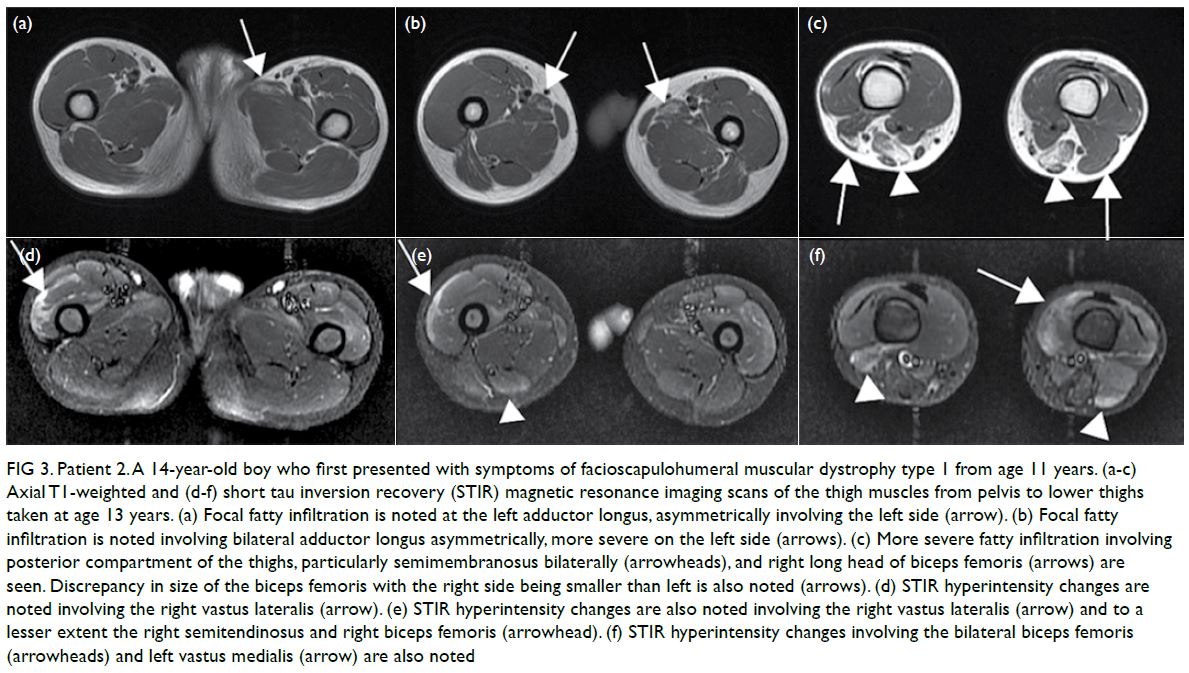
Figure 3. Patient 2. A 14-year-old boy who first presented with symptoms of facioscapulohumeral muscular dystrophy type 1 from age 11 years. (a-c) Axial T1-weighted and (d-f) short tau inversion recovery (STIR) magnetic resonance imaging scans of the thigh muscles from pelvis to lower thighs taken at age 13 years. (a) Focal fatty infiltration is noted at the left adductor longus, asymmetrically involving the left side (arrow). (b) Focal fatty infiltration is noted involving bilateral adductor longus asymmetrically, more severe on the left side (arrows). (c) More severe fatty infiltration involving posterior compartment of the thighs, particularly semimembranosus bilaterally (arrowheads), and right long head of biceps femoris (arrows) are seen. Discrepancy in size of the biceps femoris with the right side being smaller than left is also noted (arrows). (d) STIR hyperintensity changes are noted involving the right vastus lateralis (arrow). (e) STIR hyperintensity changes are also noted involving the right vastus lateralis (arrow) and to a lesser extent the right semitendinosus and right biceps femoris (arrowhead). (f) STIR hyperintensity changes involving the bilateral biceps femoris (arrowheads) and left vastus medialis (arrow) are also noted
In September 2014, Patient 3 presented with
upper and lower limb proximal muscle weakness
from age 18 years. Magnetic resonance imaging scan
was taken at age 23 years (Fig 4).
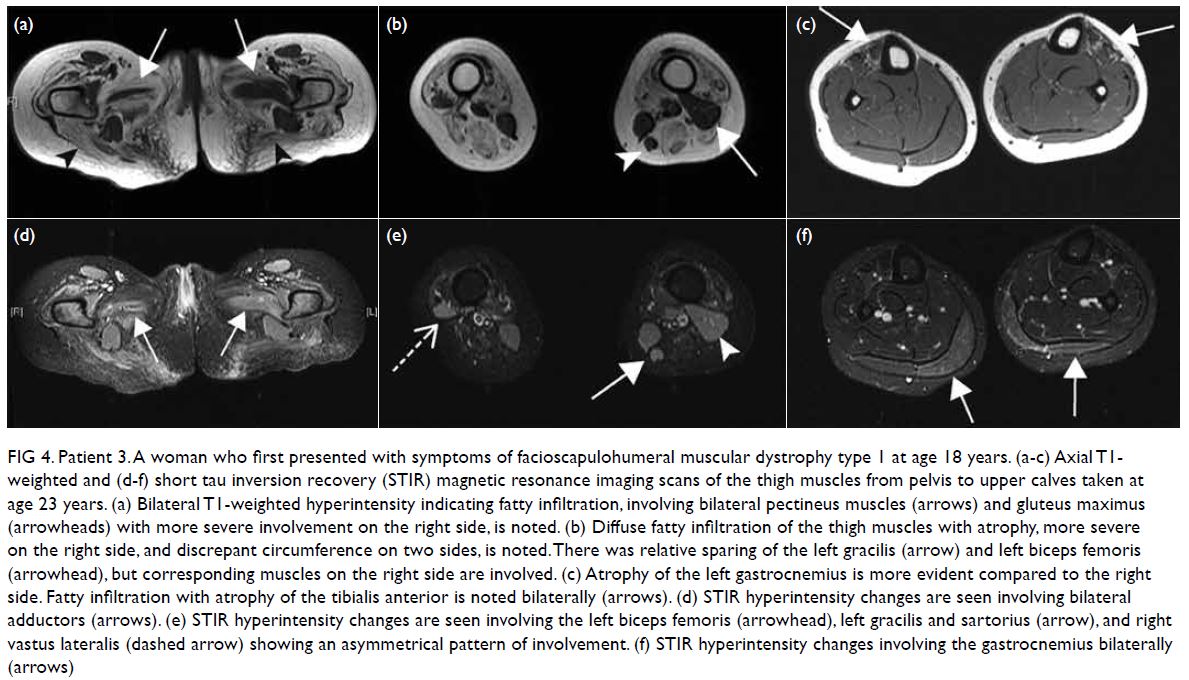
Figure 4. Patient 3. A woman who first presented with symptoms of facioscapulohumeral muscular dystrophy type 1 at age 18 years. (a-c) Axial T1- weighted and (d-f) short tau inversion recovery (STIR) magnetic resonance imaging scans of the thigh muscles from pelvis to upper calves taken at age 23 years. (a) Bilateral T1-weighted hyperintensity indicating fatty infiltration, involving bilateral pectineus muscles (arrows) and gluteus maximus (arrowheads) with more severe involvement on the right side, is noted. (b) Diffuse fatty infiltration of the thigh muscles with atrophy, more severe on the right side, and discrepant circumference on two sides, is noted. There was relative sparing of the left gracilis (arrow) and left biceps femoris (arrowhead), but corresponding muscles on the right side are involved. (c) Atrophy of the left gastrocnemius is more evident compared to the right side. Fatty infiltration with atrophy of the tibialis anterior is noted bilaterally (arrows). (d) STIR hyperintensity changes are seen involving bilateral adductors (arrows). (e) STIR hyperintensity changes are seen involving the left biceps femoris (arrowhead), left gracilis and sartorius (arrow), and right vastus lateralis (dashed arrow) showing an asymmetrical pattern of involvement. (f) STIR hyperintensity changes involving the gastrocnemius bilaterally (arrows)
Patients 2 to 8 had insidious onset of muscle
weakness during adolescence or adulthood and a
slow deterioration of motor function. Asymmetrical
muscle involvement was common. Three patients
had their lung function assessed of whom two had
a restrictive pattern suggestive of expiratory muscle
weakness.
Patients 1 and 2 were referred to the
neuromuscular disorder clinic of the Department of Paediatrics and Adolescent Medicine, The University
of Hong Kong for diagnostic examination and
testing. Patients 3 to 8 were referred to the Clinical
Genetic Service of the Department of Health for
genetic testing. All DNA diagnostic tests were
performed overseas, either self-financed (Patients
3 to 8 with the genetic testing performed in United
Kingdom) or through research collaboration with
financial support from the ‘Diagnosis and therapy
development of rare neurological diseases and
neuromuscular diseases’ fund (Patients 1 and 2, with
the genetic testing performed in The Netherlands).
For these patients, the diagnosis of FSHD was
confirmed by standard genetic testing using Southern
blotting and hybridisation with the P13E-11 probe.
Restriction enzyme digestion with EcoRI, which
recognises the D4Z4 locus on chromosome 4 and
10, was applied. The EcoRI/BlnI digestion further
fragments the chromosome 10 array to identify the
D4Z4 arrays located on chromosome 4, and the
length and number of D4Z4 units were determined.
The median age of disease onset was 14 years
(range, 1-32). The male: female ratio was 3:1. The
median time between onset of symptoms and genetic
diagnosis in this cohort was 16 years (range, 2-44). Of
the patients who required a walking stick (Patients 5
to 8), the median age at which the need arose was
47.5 years (range, 43-53). Two patients also required
a wheelchair for outdoor mobility, from age 49 and
57 years. None of the patients had hearing problems.
All eight patients with FSHD1 had a significant
contraction—from one to four units—of the D4Z4
repeats. There was no correlation between the
number of D4Z4 units and the age of onset or clinical
severity; Patients 1 and 7 both had two D4Z4 units,
but age of onset differed by 30 years.
A positive family history was observed in 38% of our patients. Patients 1 and 4 inherited the
autosomal dominant FSHD1 from their mildly
symptomatic mother, as confirmed by genetic
testing. Patient 6 inherited the condition from his
father. Patient 8 passed on FSHD1 to her daughter.
Clinical variability and reduced penetrance were
evident.
Discussion
This is the first case series of genetically confirmed
FSHD1 in Hong Kong. Worldwide, FSHD (OMIM
No. 158900) is the third most common form of
dystrophy, with a prevalence of 1:15000 to 1:20000.
It can be classified as type 1 (FSHD1) or type 2
(FSHD2). Most patients with FSHD have FSHD1
(95%) that has autosomal dominant inheritance. Up to one third of cases are caused by de novo mutations.1
Typically, FSHD presents during the second or
third decade of life as asymmetrical facial weakness
followed by muscle weakness around the scapula
and upper arms, then truncal and lower extremity
weakness. Asymmetrical muscle involvement is
typical. In all, 10% to 30% of individuals eventually become non-ambulatory. Around 38% of patients
develop a restrictive lung disease pattern, and 1% to
3% eventually require ventilatory support.
Early-onset FSHD accounts for 10% of total
FSHD. Affected individuals typically present with
facial weakness before age 5 years and shoulder
girdle weakness before age 10 years. Early-onset
FSHD is generally associated with fewer D4Z4
repeats and higher disease severity. Patients often
present with global developmental delays, dysarthria,
dysphagia, intellectual disabilities, epilepsy, cochlear
dysfunction, and retinal vasculopathy.2
The genetic mechanisms of FSHD1 are
complex (Fig 5). Genetic diagnosis of FSHD1
typically involves the Southern blot technique or the
molecular combing technique using fluorescence in
situ hybridisation.3 Presently, specific genetic testing
for FSHD1 is currently unavailable in the laboratories
of the public healthcare system in Hong Kong.
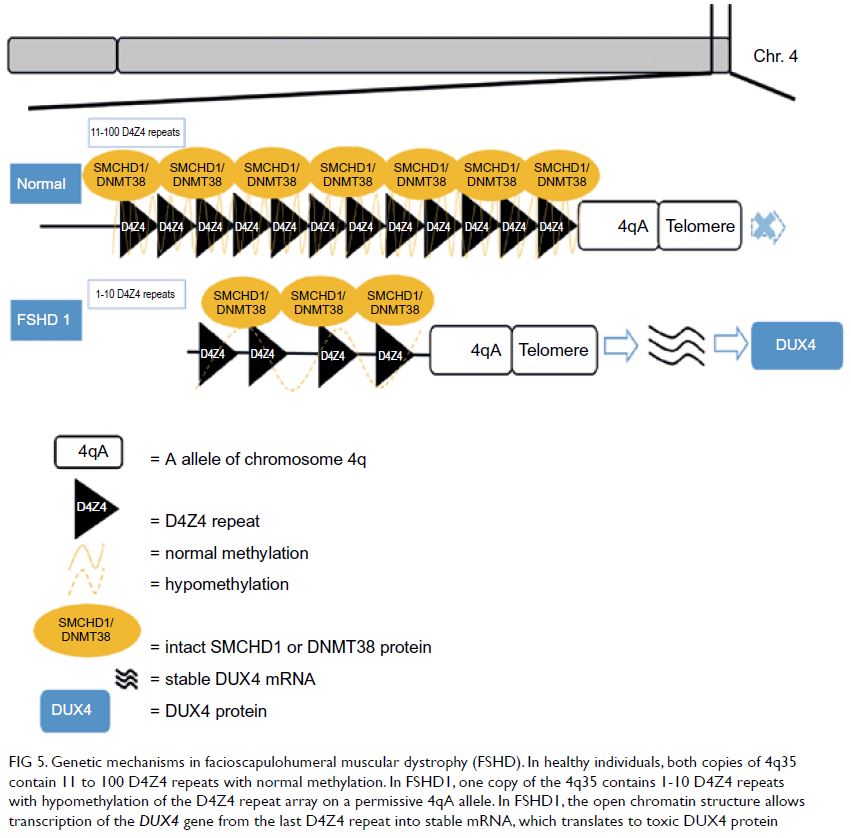
Figure 5. Genetic mechanisms in facioscapulohumeral muscular dystrophy (FSHD). In healthy individuals, both copies of 4q35 contain 11 to 100 D4Z4 repeats with normal methylation. In FSHD1, one copy of the 4q35 contains 1-10 D4Z4 repeats with hypomethylation of the D4Z4 repeat array on a permissive 4qA allele. In FSHD1, the open chromatin structure allows transcription of the DUX4 gene from the last D4Z4 repeat into stable mRNA, which translates to toxic DUX4 protein
There are several possible reasons for the delay
in diagnosis in our cases. First, the lack of patient
awareness of an underlying neuromuscular disease
at their initial presentation often led to delayed
consultation. Second, many doctors are unfamiliar
with neuromuscular diseases with consequent
delayed referrals. Most importantly, publicly funded
genetic diagnostic testing for FSHD1 is currently
unavailable. Most patients with a clinical suspicion of FSHD cannot afford expensive overseas genetic
testing to confirm their diagnosis. Some patients with
FSHD simply do not have the option of diagnosis.
Early molecular diagnosis of FSHD is crucial
to enable timely assessment and management,
including ophthalmic and hearing assessment in
early-onset FSHD; regular motor, pulmonary and
neuromuscular pain evaluations, referral for an
aerobic exercise programme, and surgical scapular
fixation if needed. Routine cardiac screening is unnecessary in the absence of cardiac symptoms.4 Early
genetic diagnosis can also help to identify other
affected or asymptomatic family members. Prenatal
diagnosis in pregnancies of affected individuals
provides couples with informed choices. Registries
for patients with FSHD have been established in
different countries to facilitate recruitment for
clinical studies and trials.
The landscape of clinical trials is promising.
A phase 2 study of losmapimod, an oral agent that
inhibits and reduces the expression of myotoxic
DUX4, is currently underway.5
This study increases professional awareness
of FSHD and highlights the importance of early
recognition and diagnosis for this condition, as well
as a current service gap in the genetic diagnosis of
FSHD. Establishing a local registry will help recruit
patients into clinical trials.
Author contributions
Concept or design: SHS Chan, WY Leung, HM Luk, V
Vardhanabhuti.
Acquisition of data: All authors.
Analysis or interpretation of data: SHS Chan, WY Leung, HM Luk, V Vardhanabhuti, Yuan Gao.
Drafting of the manuscript: SHS Chan, WY Leung.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: All authors.
Analysis or interpretation of data: SHS Chan, WY Leung, HM Luk, V Vardhanabhuti, Yuan Gao.
Drafting of the manuscript: SHS Chan, WY Leung.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take
responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Acknowledgement
The authors thank Professor Silvère M van der Maarel and Dr R Lemmers of the Department of Human Genetics, Leiden
University Medical Center, Leiden, The Netherlands, for
their expert advice on the diagnosis of facioscapulohumeral
dystrophy in the two paediatric patients. The authors also
thank Ms Rachel BY Lee for editing the English in a draft of
this manuscript.
Funding/support
This study was supported by donations from Mrs Yang to
the ‘Diagnosis and therapy development of rare neurological
diseases and neuromuscular diseases’ fund that provides
financial support for overseas genetic testing for patients with
facioscapulohumeral muscular dystrophy.
Ethics approval
This study was approved by the Hong Kong University
Institutional Review Board (Ref: UW_20-405). All patients
provided verbal consent to be included in this publication.
Two patients (Patients 1 and 2) and their families provided
written consent for the publication of clinical photographs.
References
1. Sacconi S, Salviati L, Desnuelle C. Facioscapulohumeral muscular dystrophy. Biochim Biophys Acta 2015;1852:607-14.Crossref
2. Mah JK, Chen YW. A pediatric review of
facioscapulohumeral muscular dystrophy. J Pediatr Neurol
2018;16:222-31. Crossref
3. Lemmers RJ, O’Shea S, Padberg GW, Lunt PW, van der Maarel SM. Best practice guidelines on genetic diagnostics
of Facioscapulohumeral muscular dystrophy: workshop 9th
June 2010, LUMC, Leiden, The Netherlands. Neuromuscul
Disord 2012;22:463-70. Crossref
4. Tawil R, Kissel JT, Heatwole C, et al. Evidence-based
guideline summary: Evaluation, diagnosis, and
management of facioscapulohumeral muscular dystrophy:
Report of the Guideline Development, Dissemination,
and Implementation Subcommittee of the American
Academy of Neurology and the Practice Issues Review
Panel of the American Association of Neuromuscular &
Electrodiagnostic Medicine. Neurology 2015;85:357-64. Crossref
5. Michelle Mellion M. Efficacy and safety of losmapimod in
subjects with facioscapulohumeral muscular dystrophy (FSHD). Available from: https://clinicaltrials.gov/ct2/show/NCT04003974. Accessed 2 Nov 2020.