Cardiovascular event in chronic myeloid leukaemia treated with tyrosine kinase inhibitor: a case report
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Cardiovascular event in chronic myeloid leukaemia
treated with tyrosine kinase inhibitor: a case report
YL Boo, MD, MRCP(UK); Christopher CK Liam,
MRCP(UK); SY Lim, MD, MRCP(UK); ML Look, MRCP(UK)
Department of Internal Medicine, Hospital Sultanah
Nora Ismail, Batu Pahat, Johor, Malaysia
Corresponding author: Dr YL Boo (coolrontin@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Case report
Chronic myeloid leukaemia (CML) is a
myeloproliferative neoplasm arising from a pluripotent haematopoietic stem
cell. It is associated with an oncogenic fusion gene BCR-ABL, encoding a
protein with tyrosine kinase activity.1
The emergence of tyrosine kinase inhibitors (TKIs) such as imatinib,
nilotinib, dasatinib, and ponatinib has revolutionised the treatment and
improved overall survival of patients with CML. Pericardial and pleural
effusion, pulmonary oedema, left ventricular failure, arrhythmia, and
coronary heart disease have rarely been reported in clinical trials with
nilotinib.2 We report a case of
acute coronary syndrome in a young woman treated with nilotinib.
A 33-year-old woman presented to Hospital Sultanah
Nora Ismail, Johor, Malaysia in February 2017 with first-episode
sudden-onset, left-sided chest pain that lasted more than 30 minutes and
was associated with nausea, vomiting, palpitations, and profuse
sweating. She had been diagnosed with CML 2 years previously
and had initially received imatinib with low Sokal score. Her BCR-ABL1
fusion transcript was more than 0.1% after 1 year of treatment, and thus,
her treatment was changed to nilotinib 400 mg twice daily, as a
second-line TKI. Tyrosine kinase domain mutation analysis was not
performed due to lack of resources. She was compliant with medication and
showed a good response with her BCR-ABL1 transcript dropping to less than
0.1% after 3 months of therapy. She had no other significant medical or
family history. On arrival, she was haemodynamically stable and physical
examination was normal. Her initial blood investigations showed normal
haemoglobin level (13.6 g/dL), white cell count (8.3 × 109/L),
platelet count (240 × 109/L), kidney, and liver function. Her
initial electrocardiogram showed T wave inversion over V2 to V6 with a Q
wave in lead III. Subsequent electrocardiograms showed evolving ischaemic
changes with ST depression over V2 to V6. Her initial creatine kinase was
normal (50 U/L) with negative troponin I, but rose to 950 U/L within 24
hours. Her total cholesterol was 4.2 mmol/L, low-density lipoprotein 1.6
mmol/L, and high-density lipoprotein 0.6 mmol/L. Echocardiography showed
normal ejection fraction (55%) with a dilated left atrium and left
ventricle. She was diagnosed with non–ST elevation myocardial infarction
and started on anticoagulation and dual antiplatelet therapy. Urgent
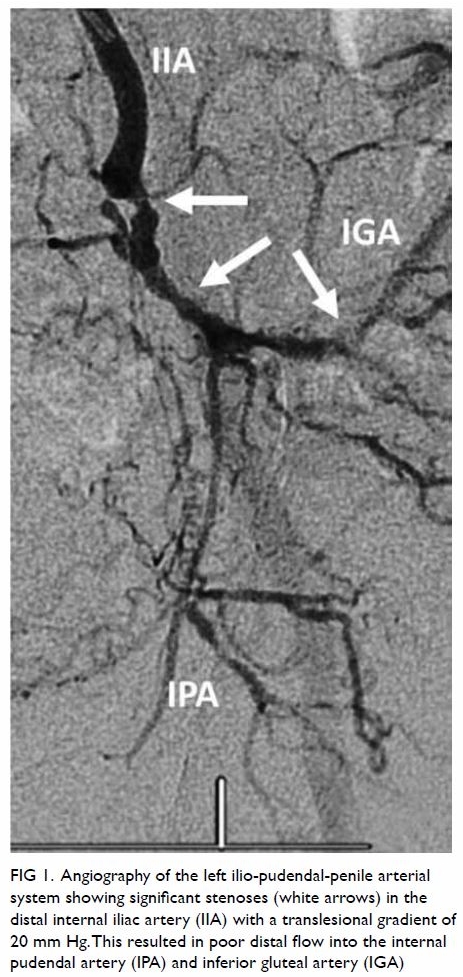
angiography showed 95% distal right coronary artery stenosis with
successful angioplasty and stenting (Fig). She was discharged from the hospital well and
planned for re-challenge of TKI during follow-up.
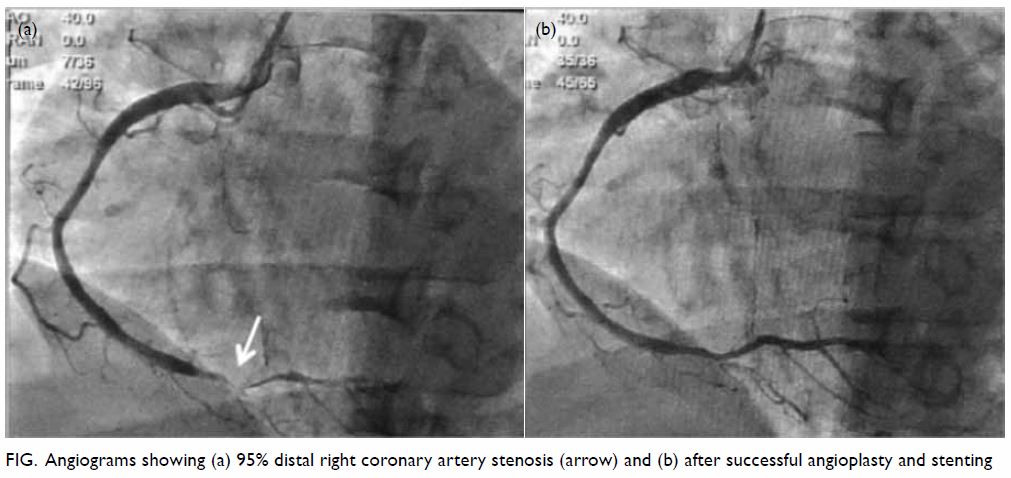
Figure. Angiograms showing (a) 95% distal right coronary artery stenosis (arrow) and (b) after successful angioplasty and stenting
Discussion
Nilotinib, a second-generation TKI, has been shown
in the ENESTnd study to induce more rapid and profound molecular responses
than imatinib in patients with CML in the chronic phase (CML-CP).2 The proxy measurements of molecular response, MR1.0,
MR2.0, MR3.0 and MR4.5 are achieved
better, and earlier on, during first-line treatment with nilotinib.2 These measurements predict better overall survival in
CML-CP,2 yet no direct proven
long-term overall survival benefit has been demonstrable for nilotinib vs
imatinib.
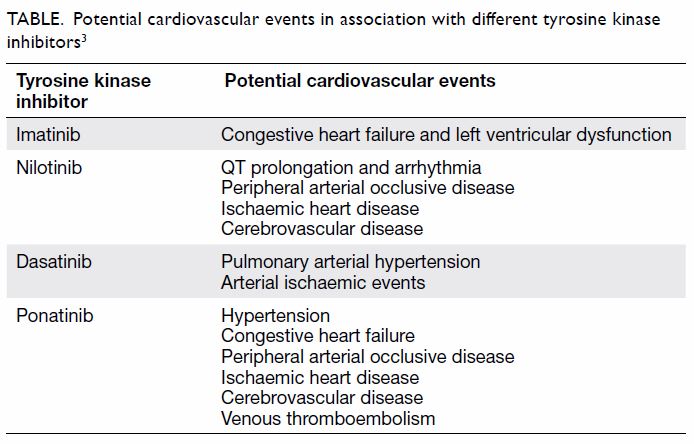
Cardiovascular adverse events with nilotinib have
raised concerns about long-term sequelae of drugs administered for decades
with 5% to 13% of patients experiencing cardiovascular events with
nilotinib.3 In addition, metabolic
effects such as hyperglycaemia, hyperlipidaemia, and increased body mass
index have significant implications for cardiovascular outcome in patients
treated with nilotinib.2 4 Preliminary studies suggest nilotinib has detrimental
effects on endothelial cell function in vitro and may accelerate
atherosclerosis in addition to the metabolic effects.5 Therefore, cardiovascular risk assessment needs to be
integrated and regular monitoring is important especially in patients at
high risk of cardiac disease. Our patient presented with acute coronary
syndrome after 1 year of treatment with nilotinib. She was previously
taking imatinib, but current evidence suggests a lower incidence of
cardiovascular events in patients taking imatinib, even compared with
those not taking TKIs (Table).3
Early cardiac intervention and optimisation of risk factors may improve
overall morbidity and mortality.
In summary, cardiac events have been reported in
CML patients treated with nilotinib. Therefore, it is important to
recognise these possible complications. Early treatment can then be
instituted to improve overall outcome.
Author contributions
All authors contributed to the concept, acquisition
of data, analysis of data, drafting of the manuscript, and critical
revision of important intellectual content. All authors had full access to
the data, contributed to the study, approved the final version for
publication, and take responsibility for its accuracy and integrity.
Acknowledgements
The authors would like to thank the Director
General of Health Malaysia for permission to publish this article.
Conflicts of interest
All authors have disclosed no conflicts of
interest.
Funding/support
This paper received no specific grant from any
funding agency in the public, commercial, or not-for-profit sectors.
Patient consent
The patient provided written consent for
publication.
References
1. Vardiman JW, Melo JV, Baccarani M,
Thiele J. Chronic myelogenous leukaemia, BCR-ABL1 positive. In: Swerdlow
SH, Campo E, Harris NL, et al, editors. WHO Classification of Tumours of
Haematopoietic and Lymphoid Tissues. 4th ed. Lyon: IARC; 2008: 32-7.
2. Saglio G, Kim DW, Issaragrisil S, et al.
Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N
Engl J Med 2010;362:2251-9. Crossref
3. Moslehi JJ, Deininger M. Tyrosine kinase
inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia.
J Clin Oncol 2015;33:4210-8. Crossref
4. Rea D, Mirault T, Cluzeau T, et al.
Early onset hypercholesterolemia induced by the 2nd-generation tyrosine
kinase inhibitor nilotinib in patients with chronic phase-chronic myeloid
leukemia. Haematologica 2014;99:1197-203. Crossref
5. Emir H, Albrecht-Schgoer K, Huber K, et
al. Nilotinib exerts direct pro-atherogenic and anti-angiogenic effects on
vascular endothelial cells: a potential explanation for drug-induced
vasculopathy in CML. Blood 2013;122:257.