A lucky and reversible cause of 'ischaemic bowel'
DOI: 10.12809/hkmj144306
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
A lucky and reversible cause of ‘ischaemic bowel’
YF Shea, MRCP, FHKAM (Medicine)1;
Felix CL Chow, MB, BS, MRCS2;
F Chan, MRCP, FHKAM (Medicine)1;
Janice JK Ip, MB, BS, FRCR3;
Patrick KC Chiu, FRCP(Glasg), FHKAM (Medicine)1;
Fion SY Chan, FRCS, FHKAM (Surgery)2;
LW Chu, MD, FRCP1
1 Acute Geriatric Unit, Grantham Hospital, Aberdeen, Hong Kong
2 Department of Surgery, Queen Mary Hospital, The University of Hong
Kong, Pokfulam, Hong Kong
3 Department of Radiology, Queen Mary Hospital, The University of Hong
Kong, Pokfulam, Hong Kong
Corresponding author: Dr YF Shea (elphashea@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
An 81-year-old man was admitted with an infective
exacerbation of chronic obstructive pulmonary
disease. He also had clinical and radiological features
suggestive of ileus. On day 6 after admission, he
developed generalised abdominal pain. Urgent
computed tomography of the abdomen showed
presence of portovenous gas and dilated small
bowel with pneumatosis intestinalis and whirl
sign. Emergency laparotomy was performed, which
showed a 7-mm perforated ulcer over the first part
of the duodenum and small bowel volvulus. Omental
patch repair and reduction of small bowel volvulus
were performed. No bowel resection was required.
The patient had a favourable outcome. Clinicians
should suspect small bowel volvulus as a cause of
ischaemic bowel. Presence of portovenous gas and
pneumatosis intestinalis are normally considered to
be signs of frank ischaemic bowel. The absence of
bowel ischaemia at laparotomy in this patient shows
that this is not necessarily the case and prompt
surgical treatment could potentially save the bowels
and lives of these patients.
Introduction
Ischaemic bowel is often associated with high
mortality. Common causes of ischaemic bowel
include atrial fibrillation with arterial embolism,
incarcerated hernia, volvulus, profound shock, and
vasculitis. Volvulus in adults most often occurs
in the colon and sigmoid colon (70-80%), and
less commonly in caecum (10-20%).1 Small bowel
volvulus (SBV) is rarely encountered in adults.1 2 3 4 5 6 7 8 9
Intra-operatively, if gangrenous small bowel is found,
it needs to be resected and may potentially cause
long-term complications, including short bowel
syndrome.2 We report on a patient with primary SBV
associated with a perforated duodenal ulcer (PDU),
but the patient had a favourable outcome without
the need for bowel resection, thereby avoiding the
long-term complications associated with extensive
bowel resection.
Case report
In February 2014, an 81-year-old man was admitted
with an infective exacerbation of chronic obstructive
pulmonary disease caused by influenza B virus.
He had a history of left inguinal hernia repair. On
admission, he had a temperature of 38°C. He was
tachypnoeic with diffuse expiratory wheeze on chest
auscultation. His abdomen was slightly distended
and bowel sounds were sluggish. There was no
recurrence of the hernia. Complete blood count
showed leukocytosis (23.7 x 109 /L [reference range,
4.5-11.0 x 109 /L]) and a haemoglobin level of 109 g/L
(reference range, 140-175 g/L). Liver and renal
function tests and electrolytes were normal. Chest
radiograph showed hyperinflation of the lungs with
no consolidation. Abdominal radiograph showed
dilated small bowel without air-fluid level.
The patient was treated with inhaled
bronchodilators (albuterol 4 puffs every 4 hours
and ipratropium bromide 2 puffs every 4 hours) and
intravenous amoxicillin-clavulanate 1.2 g every 8
hours for 6 days. The dilated small bowel was initially
attributed to ileus and the patient was kept nil-by-mouth.
Daily abdominal radiograph did not reveal
any worsening of the small bowel dilatation. He
developed an episode of fast atrial fibrillation on day
2, which was controlled with amiodarone (initially
with intravenous loading and infusion, then followed
by 200 mg orally twice a day). Flatus and bowel
opening returned on day 5 and diet was resumed. On
day 6, the patient developed generalised abdominal
pain. His abdomen was distended with no localised
tenderness, but bowel sounds were absent. Arterial
blood gas showed no metabolic acidosis. Computed
tomography (CT) of the abdomen with contrast
showed presence of portovenous gas, dilated small
bowel with pneumatosis intestinalis, and whirl sign
(Fig 1).
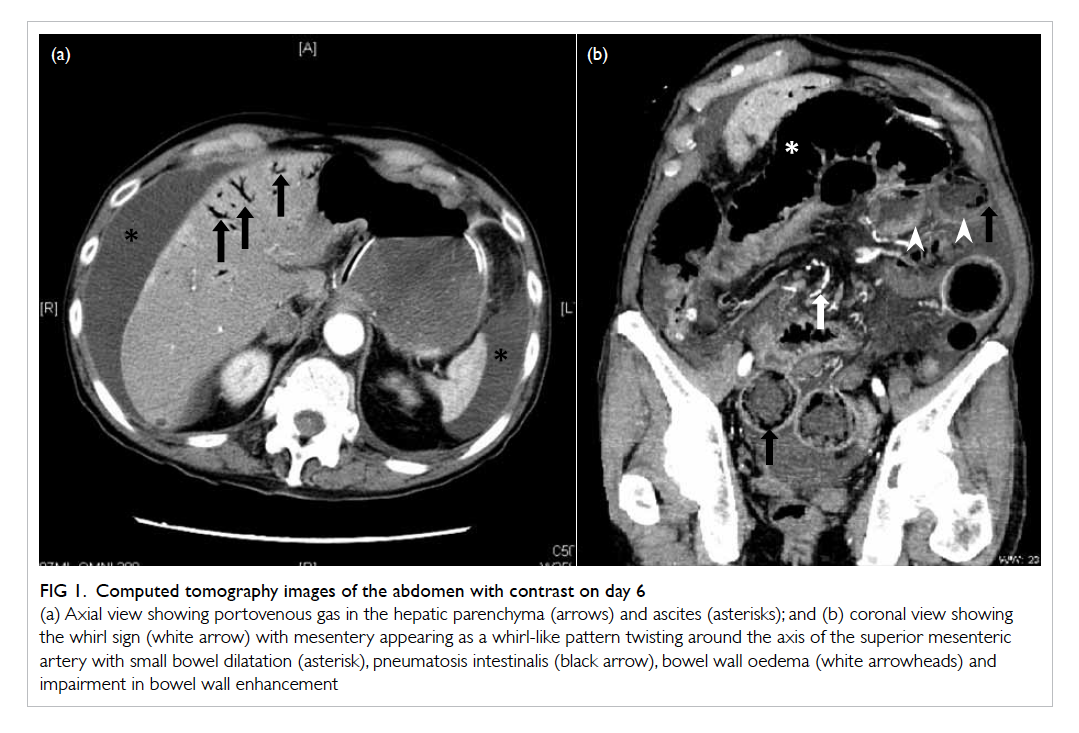
Figure 1. Computed tomography images of the abdomen with contrast on day 6
(a) Axial view showing portovenous gas in the hepatic parenchyma (arrows) and ascites (asterisk); and (b) coronal view showing the whirl sign (white arrow) with mesentery appearing as a whirl-like pattern twisting around the axis of the superior mesenteric artery with small bowel dilatation (asterisk), pneumatosis intestinalis (black arrow), bowel wall oedema (white arrowheads) and impairment in bowel wall enhancement
Emergency laparotomy was performed that
showed a 7-mm perforated ulcer over the first
part of the duodenum, which was sealed off by the
adjacent liver and falciform ligament. The small
bowel was grossly distended. There was SBV with
twisting of the small bowel along the root of the
mesentery by 180° with mild dusky appearance.
Omental patch repair of the duodenal ulcer and
reduction of the SBV were performed. Small bowel
perfusion improved significantly and a strong
superior mesenteric artery pulse was confirmed.
Bowel resection was not required. Histological
examination of the duodenal ulcer showed no
evidence of Helicobacter pylori infection. Repeated
CT of the abdomen on day 8 showed resolution of
portovenous gas (Fig 2). Postoperatively, the patient
had the complication of an intra-abdominal abscess,
which was managed successfully by percutaneous
drainage and systemic antibiotics (vancomycin
500 mg daily and meropenem 1 g every 12 hours
intravenously for 2 weeks followed by levofloxacin
500 mg orally daily for 1 week). He was discharged
on day 36.
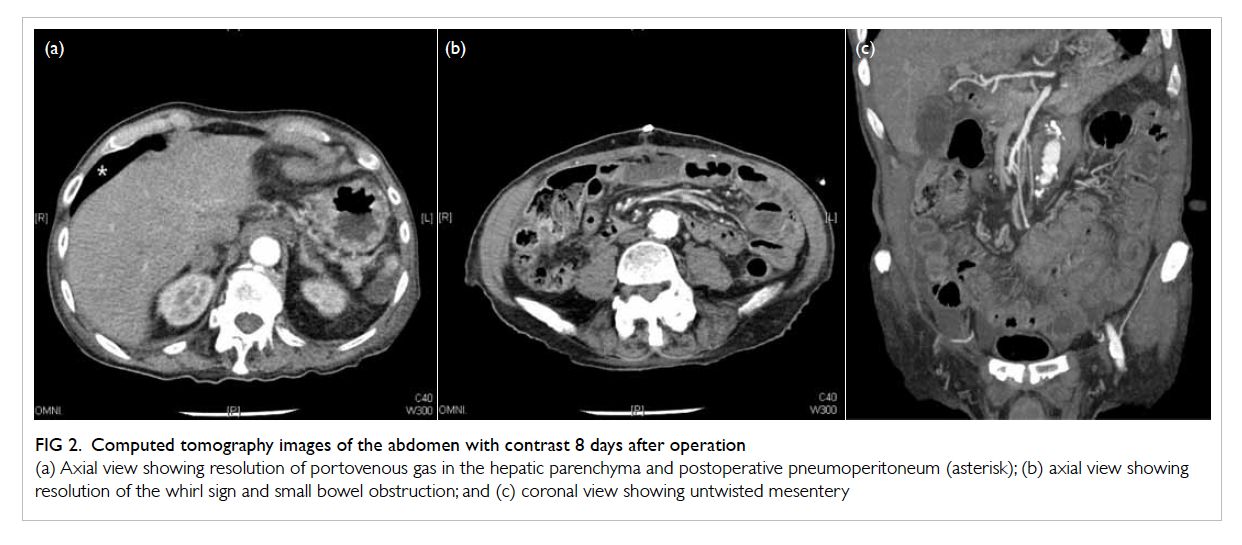
Figure 2. Computed tomography images of the abdomen with contrast 8 days after operation
(a) Axial view showing resolution of portovenous gas in the hepatic parenchyma and postoperative pneumoperitoneum (asterisk); (b) axial view showing resolution of the whirl sign and small bowel obstruction; and (c) coronal view showing untwisted mesentery
Discussion
The initial clinical feature of dilated small bowel
with sluggish bowel sound in the background of
an infective exacerbation of chronic obstructive
pulmonary disease was interpreted as being caused
by paralytic ileus.10 With the sudden worsening in
abdominal pain and the presence of paroxysmal
atrial fibrillation, the possibility of ischaemic
bowel was reconsidered. This explains why SBV
was only diagnosed by CT of the abdomen on day
6 after admission. Small bowel volvulus refers to
the twisting of a small bowel segment around the
axis of its own mesentery. In the elderly patients,
a secondary type with an underlying cause is
most commonly encountered.3 These secondary
causes include tumours, mesenteric lymph nodes,
adhesions after previous surgery, malrotation,
congenital bands, intussusception, colostomy, and
internal hernias.3 If no underlying cause is found, as
in this patient, the SBV is considered to be a primary
SBV (some surgeons may regard malrotation and
congenital adhesions as primary SBV).3 4 There are
certain risk factors that have been linked to primary
SBV, including long mobile mesentery, short
mesenteric base, long small bowel, and consumption
of a large amount of fibre-rich food after prolonged
fasting, with overloading of an empty intestine.1 3 6
With torsion of the small bowel and its mesentery,
the superior mesenteric arterial blood supply is
compromised, which results in bowel infarction that
usually occurs within 6 hours.3
The most important clinical feature is
persistent abdominal pain with absence of bowel
sounds on physical examination.1 2 3 4 5 6 7 8 9 Plain abdominal
radiography is of limited diagnostic value.4
Abdominal CT is the most useful tool for obtaining
the correct preoperative diagnosis.4 The findings
on CT of the abdomen reflect the underlying
pathophysiology of the SBV. Whirl sign, which
refers to the twisting of mesentery around the
origin of the torsion, is the most typical radiological
sign of SBV.4 7 8 9 Alternatively, a venous cut-off sign,
referring to the occlusion of the superior mesenteric
vein, may also be found.9 With lymphatic and
venous occlusions, there will be small bowel wall
oedema and thickening.4 Small bowel dilatation is
caused by intestinal obstruction resulting from the
torsion.4 The bowel wall ischaemia results from the
pneumatosis intestinalis and portovenous gas.4 It has
been proposed that with intestinal ischaemia, the gas
produced by gas-forming organisms accumulates
in the bowel wall (pneumatosis intestinalis) and
circulates to the liver (portovenous gas); alternatively
the gas-producing organisms may enter the portal
circulation and produce the gas there (portovenous
gas).11
The causal relationship between SBV and
PDU in this patient is not well established. We
suspected that both the SBV and a non-PDU might
be present during the early phase of admission.
The resumption of a meal after a period of fasting
exacerbated the volvulus. It has been proposed that
the transit of food contents into the empty proximal
jejunum could cause a gravitational migration of this
segment into the left lower quadrant; forcing the
distal empty bowel loops in a clockwise direction
to the right upper quadrant with torsion of the
mesentery.6 The mechanical obstruction contributes
to the perforation of the duodenal ulcer which was a
weak point and, together with the impending bowel
ischaemia, led to the generalised abdominal pain.
The possibility of ileus converting into SBV was less
likely because of the usually reversible nature of ileus
with treatment of the infection.
A favourable outcome for patients with SBV
depends on prompt emergency laparotomy.1 2 3 4 5 6 7 8 9
The absence of bowel gangrene intra-operatively
predicted a favourable outcome for this patient.
Strictly, volvulus is defined by more than 180° of
rotation, so the intra-operative finding of small bowel
twisting around the mesentery root by 180° may
signify that the SBV was in the process of partially
reversing by itself. In one case series involving 19
patients, Ruiz-Tovar et al6 noted 100% mortality in
patients with an intra-operative finding of bowel
wall gangrene. Birnbaum et al3 also reported the use
of enteropexy to prevent recurrence of primary SBV
in a 69-year-old man with long mobile mesentery.
In summary, clinicians should suspect SBV as
a cause of ischaemic bowel. Presence of portovenous
gas and pneumatosis intestinalis are normally
considered signs of frank ischaemic bowel. The
absence of bowel infarction in this patient illustrates
that this is not necessarily the case and prompt
surgical treatment could potentially save the bowels
and lives of these patients.
References
1. Kim KH, Kim MC, Kim SH, Park KJ, Jung GJ. Laparoscopic
management of a primary small bowel volvulus: a case
report. Surg Laparosc Endosc Percutan Tech 2007;17:335-8. Crossref
2. Rubio PA, Galloway RE. Complete jejunoileal necrosis due
to torsion of the superior mesenteric artery. South Med J
1990;83:1482-3. Crossref
3. Birnbaum DJ, Grègoire E, Campan P, Hardwigsen J, Le
Treut YP. Primary small bowel volvulus in adult. J Emerg
Med 2013;44:e329-30. Crossref
4. Jaramillo D, Raval B. CT diagnosis of primary small-bowel
volvulus. AJR Am J Roentgenol 1986;147:941-2. Crossref
5. Weledji EP, Theophile N. Primary small bowel volvulus in
adults can be fatal: a report of two cases and brief review of
the subject. Trop Doct 2013;43:75-6. Crossref
6. Ruiz-Tovar J, Morales V, Sanjuanbenito A, Lobo E,
Martinez-Molina E. Volvulus of the small bowel in adults.
Am Surg 2009;75:1179-82.
7. Li CH, Chen CH, Chou JW. Intestinal obstruction caused
by small bowel volvulus. Am J Med 2011;124:e3-4. Crossref
8. Huang YM, Wu CC. Whirl sign in small bowel volvulus.
BMJ Case Rep 2012;2012:bcr2012006688.
9. Ho YC. “Venous cut-off sign” as an adjunct to the “whirl
sign” in recognizing acute small bowel volvulus via CT
scan. J Gastrointest Surg 2012;16:2005-6. Crossref
10. Batke M, Cappell MS. Adynamic ileus and acute colonic
pseudo-obstruction. Med Clin North Am 2008;92:649-70,ix. Crossref
11. Abboud B, El Hachem J, Yazbeck T, Doumit C. Hepatic
portal venous gas: physiopathology, etiology, prognosis
and treatment. World J Gastroenterol 2009;15:3585-90. Crossref