DOI: 10.12809/hkmj154604
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
A case of refractory seizure with cognitive impairment due to anti-GABA encephalitis
Adrian TH Hui, MB BS, MRCP (UK); YO Lam, MB, BS, MRCP (UK); CK Chan, MB, BS, FHKAM (Medicine); KY Cheung, MB, ChB, FHKAM (Medicine); BH Fung, MB, BS, FHKAM (Medicine); PW Ng, MB, BS, FHKAM (Medicine)
Department of Medicine and Geriatrics, United Christian Hospital, Kwun Tong, Hong Kong
Corresponding author: Dr Adrian TH Hui (hth077@ha.org.hk)

Case report
A 57-year-old man presented with first episode
of loss of consciousness at work in February 2014.
He experienced no symptoms prior to this syncope
event. He was witnessed by his colleagues to have
tonic rigidity over his upper limbs, with a bite mark
evident over the lateral aspect of his tongue. He
had experienced a right-sided temporal headache
characterised as dull and persistent for 3 weeks prior
to this incident without any precipitating factors or
aura identified.
He enjoyed good past health and had been
educated to high school level. He was currently
working as an accountant. He did not smoke or
drink, had no recent travel history, and was not on
any long-term medication or taking herbs. He could
not recall any febrile convulsion during childhood or
significant family history of neurological diseases.
He was fully oriented and remained afebrile on
admission. Routine blood tests (electrolytes, glucose,
and inflammatory markers), physical examination,
and initial computed tomographic (CT) brain were
unremarkable. The patient refused lumbar puncture
and was discharged against medical advice.
Three days after discharge, he was admitted
to a private hospital with generalised tonic-clonic
convulsion. Electroencephalography was normal
and magnetic resonance imaging (MRI)/magnetic
resonance angiography brain showed no mass lesion
or infarct. He was discharged with a prescription
of levetiracetam in view of the second episode of
seizure.
He was again admitted to us in early March
2014 with breakthrough seizure and post-ictal
drowsiness, despite good drug compliance and no
identifiable precipitating factors. His seizure was
initially controlled with levetiracetam with the
addition of phenytoin, but he remained disoriented
with confused speech. His Mini-Mental State
Examination (MMSE) score was 14/30: his main
deficit was delayed recall with failure to perform serial sevens and to copy a polygon. He developed a low-grade
fever of 37.8°C with negative septic workup. Lumbar
puncture did not reveal any evidence of central
nervous system infection (white cell count, <1/mm3,
protein level not elevated, and negative culture
result). Electroencephalography showed right
temporal spikes only. He was prescribed empirical
Augmentin (Sandoz, Australia) 1.2 g every 8 hours
intravenously and his fever settled.
One week after admission, he experienced
multiple episodes of generalised tonic-clonic
seizure without progression to status epilepticus.
Doses of both phenytoin and levetiracetam were
increased along with the addition of valproic
acid. His seizures abated but cognition was not
improved. Electroencephalography was repeated and
demonstrated left temporal polyspikes over F7/T3/T5
in addition to bitemporal sharp waves (Fig 1). Brain
MRI was also repeated and showed no abnormality
(Fig 2). Blood tests were unremarkable with normal
electrolytes, negative tumour markers (alpha-fetoprotein/carcinoembryonic antigen/prostate-specific
antigen), negative autoimmune markers
(antinuclear antibodies/rheumatoid factor), normal
thyroid function and lactate level, and human immunodeficiency virus–negative
status. Lumbar puncture was repeated, and this
time showed mild lymphocytic pleocytosis with
white cell count of 9/mm3 (98% lymphocyte count),
protein level again not elevated, cerebrospinal fluid
(CSF)–serum glucose ratio of 0.53 (4.1/7.8). Both serum and CSF
oligoclonal bands were present, and immunoglobulin
G index was also elevated.
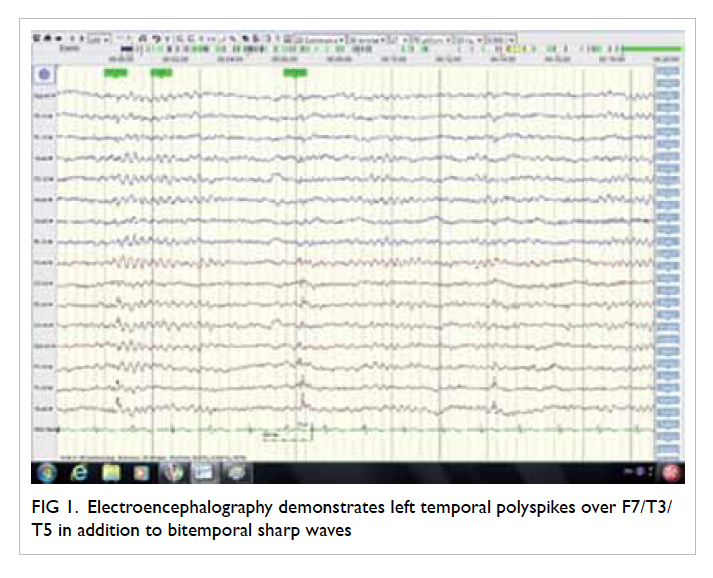
Figure 1. Electroencephalography demonstrates left temporal polyspikes over F7/T3/T5 in addition to bitemporal sharp waves
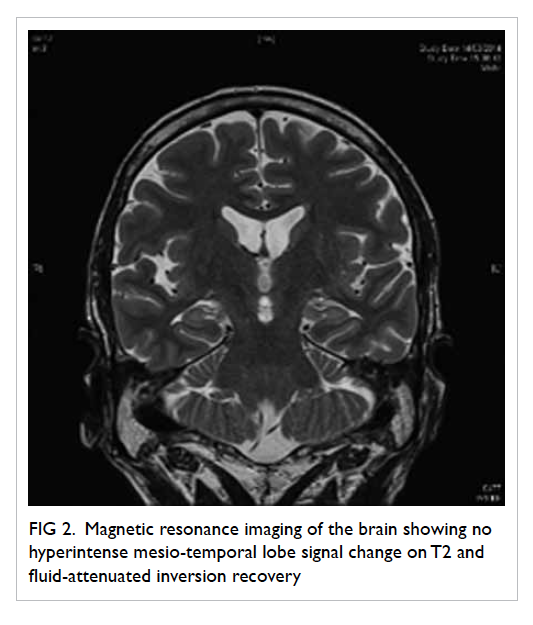
Figure 2. Magnetic resonance imaging of the brain showing no hyperintense mesio-temporal lobe signal change on T2 and fluid-attenuated inversion recovery
Both autoimmune encephalitis and post-ictal
pleocytosis are important differential diagnoses
to be considered. Serum and CSF were saved for
the detection of auto-antibodies, although anti–N-methyl
D-aspartate (anti-NMDA), anti–voltage-gated
potassium channel (anti-VGKC), and antineuronal
(anti-Hu/anti-Ri/anti-Yo) antibodies were
all negative, anti-GABAB antibodies were detected
in both serum and CSF of this patient.
Discussion
Autoimmune encephalitis, previously labelled
limbic encephalitis, was first described in the 1960s
when patients with lung cancer also suffered from
temporal lobe epilepsy, memory loss, and dementia
features.1 It was once thought that limbic encephalitis
was always associated with malignancy. In 1985, the
first onconeuronal antibody, anti-Hu antibody, was
discovered in small-cell lung carcinoma patients.1
More antibodies were identified in subsequent
years, namely CV2/CRMP5-Ab and Ma2-Ab, that
target intracellular peptides being expressed on
the cell membrane.1 At the turn of the 21st century,
patients who had features of limbic encephalitis
did not necessarily develop malignancy, and those
affected were usually young individuals who
responded well to immunotherapy. Researchers
later found antibodies in these patients that targeted
cell membrane antigens that are receptors involved
in synaptic transmission, plasticity, and neuronal
excitability. The first such antibody discovered was
VGKC.1
The pathophysiological mechanism involves
both humeral and cellular immunity mediated
by antibody production and cytotoxic T cells,
respectively.1 The hippocampus and hypothalamus
are most vulnerable during the active disease stage
as the permeability of the blood-brain barrier is
increased more than other regions of the brain, and
is thereby more susceptible to autoimmune attack.2
Treatment modalities include removal of the
underlying tumour and the antibodies. Thus far,
no randomised controlled trials have analysed the
efficacy of such treatment. Clinical experience would
suggest that the first-line immunotherapy would be
steroid as anti-inflammatory agent and intravenous
immunoglobulin (IVIG)/plasma exchange for
antibody removal.3 If first-line treatment becomes
ineffective, second-line treatment including
rituximab as a B-cell depleting agent and
cyclophosphamide as an anti-inflammatory agent
would be considered.2
As clinicians, it is important to recognise classic
features that alert us to the possibility of autoimmune
encephalitis. These features include subacute onset
of confusion, short-term memory loss, behaviour
change (depression, apathy, irritability), and seizures
(usual temporal complex partial type).4 These
features can precede the development of cancer
for paraneoplastic encephalitis.5 It is important to
obtain the history of smoking and family history of
malignancy. The occurrence of refractory seizure
despite prescription of multiple antiepileptics should
prompt the clinician to consider this diagnosis as well.
As part of the workup to exclude other differential
diagnoses, lumbar puncture, electroencephalogram,
and brain MRI should be performed, although these
are non-specific tests for confirming autoimmune
encephalitis.4 A CSF picture of lymphocytosis with
mildly elevated protein and presence of oligoclonal
bands will be present.4 Electroencephalography
will show temporal lobe abnormalities, while brain
MRI will reveal a unilateral or bilateral hyperintense
medial temporal lobe signal change on T2 and fluid-attenuated
inversion recovery without contrast
enhancement that can progress to hippocampal and
temporal lobe atrophy.4 6
A condition that mimics autoimmune
encephalitis is infectious encephalitis, particularly
herpes encephalitis. To differentiate the two, several
clinical clues might be useful. Fever is almost always
present in infectious encephalitis; it is present in
about 50% of autoimmune encephalitis cases.6 Skin
lesions can be found in varicella-zoster infection.6
Lymphocytic pleocytosis is milder in autoimmune
encephalitis than in viral illnesses.6 Periodic lateral
epileptic discharge over the temporal region can
be found in herpes encephalitis, and typical brain
MRI findings in this aetiology would be asymmetric
medial temporal lobe necrosis along with cingulate
and insular region involvement.6 Herpes simplex
virus–polymerase chain reaction in CSF would be
essential as part of the workup as well. Ultimately,
for definitive diagnosis of autoimmune encephalitis,
paired serum and CSF antibodies should be obtained.
In GABAB encephalitis, antibodies target
both GABAB1 and B2 subunits located mainly
in the hippocampus, thalamus, and cerebellum.7
Patients usually have early prominent seizures
(temporal lobe epilepsy), memory deficits, increased
anxiety, and mood dysregulation. Novel symptoms
such as ataxia or opsoclonus-myoclonus have also
been recently reported.7 8 Small-cell lung carcinoma is often present.7 Concurrent antibodies such as
VGKC-Ab, GAD-Ab (glutamic acid decarboxylase)
have also been documented.7 9 The outcome is driven by the adequacy of tumour removal if found and
the presence of other auto-antibodies, particularly
onconeural antibodies (amphiphysin and SOX1)
that are associated with a poorer prognosis.8
In a recent literature review, the discovery of
anti-GABAA antibody as a target of autoimmunity
has received much attention as a cause of refractory
seizures or status epilepticus.10 In contrast to
GABAB encephalitis, extensive cortical-subcortical
brain MRI abnormalities and the co-existence
with other antibodies particularly GAD65 or TPO
(thyroid peroxidase) were shown in a recent case
series report.10
To monitor disease activity, both serum and
CSF antibodies should be collected as some are more
readily detectable in one compartment than another:
NMDA antibodies are readily obtained from CSF and
VGKC from serum.2 In general, treatment such as
long-term immunosuppression should be guided by
clinical judgement and not necessarily on antibody
level.
In our patient, after a 5-day course of
IVIG there was no recurrence of seizure. Oral
prednisolone (1 mg/kg/day) was then prescribed and
slowly tapered. His cognition was assessed 1 week
after IVIG. Although MMSE remained low with a
score of 15/30, improvement in abstract thinking,
calculation, and proverb interpretation was noted
upon discharge.
Positron emission tomography/CT was
performed in late March 2014 to screen for
malignancy. There was abnormal fluorodeoxyglucose
uptake over the medial aspect of the left temporal
lobe. Brain MRI was repeated 2 months after
discharge and showed resolved signal/swelling in the
medial aspect of the left temporal lobe. His cognition
was again assessed in November 2014 and showed
improvement in orientation and judgement, but still
moderately to severely impaired memory.
Declaration
No conflict of interests was declared by the authors.
Acknowledgement
I would like to thank Dr Josep Dalmau, Service of
Neurology, Hospital Clinic, University of Barcelona,
Spain, for his assistance in processing our sample.
References
1. Didelot A, Honnorat J. Autoimmune limbic encephalitis.
Future Neurol 2011;6:97-111. Crossref
2. Vincent A, Bien CG, Irani SR, Waters P. Autoantibodies
associated with diseases of the CNS: new developments
and future challenges. Lancet Neurol 2011;10:759-72. Crossref
3. Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment
and prognostic factors for long-term outcome in patients
with anti-NMDA receptor encephalitis: an observational
cohort study. Lancet Neurol 2013;12:157-65. Crossref
4. Derry CP, Wilkie MD, Al-Shahi Salman R, Davenport
RJ. Autoimmune limbic encephalitis. Clin Med (Lond)
2011;11:476-8. Crossref
5. Dalmau J, Rosenfeld MR. Paraneoplastic syndromes of the
CNS. Lancet Neurol 2008;7:327-40. Crossref
6. Armangue T, Leypoldt F, Dalmau J. Autoimmune
encephalitis as differential diagnosis of infectious
encephalitis. Curr Opin Neurol 2014;27:361-8. Crossref
7. Lancaster E, Lai M, Peng X, et al. Antibodies to the
GABAB receptor in limbic encephalitis with seizures:
case series and characterisation of the antigen. Lancet
Neurol 2010;9:67-76. Crossref
8. Höftberger R, Titulaer MJ, Sabater L, et al. Encephalitis and
GABAB receptor antibodies: novel findings in a new case
series of 20 patients. Neurology 2013;81:1500-6. Crossref
9. Boronat A, Sabater L, Saiz A, Dalmau J, Graus F. GABAB
receptor antibodies in limbic encephalitis and anti-GAD–associated
neurologic disorders. Neurology 2011;76:795-800. Crossref
10. Petit-Pedrol M, Armangue T, Peng X, et al. Encephalitis
with refractory seizures, status epilepticus, and antibodies
to the GABAA receptor: a case series, characterisation of
the antigen, and analysis of the effects of antibodies. Lancet
Neurol 2014;13:276-86. Crossref