Clozapine-induced acute interstitial nephritis
DOI: 10.12809/hkmj144312
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Clozapine-induced acute interstitial nephritis
SY Chan, MRCP (UK), FHKCP1;
CY Cheung, PhD, FHKCP1;
PT Chan, MB, BS, FHKCPath2;
KF Chau, FHKCP, FRCP (Lond)1
1 Department of Medicine, Queen Elizabeth Hospital, Jordan, Hong Kong
2 Department of Pathology, Queen Elizabeth Hospital, Jordan, Hong Kong
Corresponding author: Dr SY Chan (helenchansy@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
Acute interstitial nephritis is a common cause of
acute kidney injury. Acute interstitial nephritis is
most commonly induced by drug although the cause
may also be infective, autoimmune, or idiopathic.
Although eosinophilia and eosinophiluria may help
identify this disease entity, the gold standard for
diagnosis remains renal biopsy. Prompt diagnosis
is important because discontinuation of the culprit
drugs can reduce further kidney injury. We present a
patient with an underlying psychiatric disorder who
was subsequently diagnosed with clozapine-induced
acute interstitial nephritis. Monitoring of renal
function during clozapine therapy is recommended
for early recognition of this rare side-effect.
Case report
A 29-year-old woman with a known history
of bipolar affective disorder and paranoid
schizophrenia admitted to our hospital because of
relapse of psychiatric symptoms in June 2011. She
had no significant medical illness. Her medication
included quetiapine fumarate and sodium valproate
but was changed to haloperidol decanoate depot
injection, trihexyphenidyl, and clozapine following
hospitalisation. Clozapine was commenced at 50 mg
once per day and gradually stepped up to 400 mg once
per day. One week later she developed fever but had
no respiratory or urinary symptoms. She remained
conscious and alert with stable vital signs. Physical
examination revealed no significant abnormality and
preliminary investigations showed normochromic
and normocytic anaemia with haemoglobin level of
87 g/L, white cell count of 11.8 x 109 /L (eosinophils
15.1%; absolute count 2.3 x 109 /L), and serum
creatinine of 229 µmol/L (baseline serum creatinine
1 week ago, 39 µmol/L). Her liver enzymes, lactate
dehydrogenase, and haptoglobin were all within
normal range. Chest radiograph showed clear
lung fields. She was empirically given amoxicillin/clavulanic acid but treatment was complicated by
gastro-intestinal side-effects such as vomiting and
diarrhoea. The antibiotics were stopped immediately
and the symptoms subsided. Clozapine was further
titrated up to 700 mg once per day with consequent
improvement in her mental state. Nonetheless,
fever persisted with further deterioration in renal
function (serum creatinine rose to 356 µmol/L).
Autoimmune markers including anti–nuclear
antibody, anti–neutrophil cytoplasmic antibodies,
and anti–glomerular basement membrane antibody
were all within normal range. Urinalysis revealed
the presence of red blood cells and eosinophils
but urine culture showed no bacterial growth. The
24-hour urine protein was 1.18 g. Ultrasound of the
urinary system showed normal-sized kidneys with
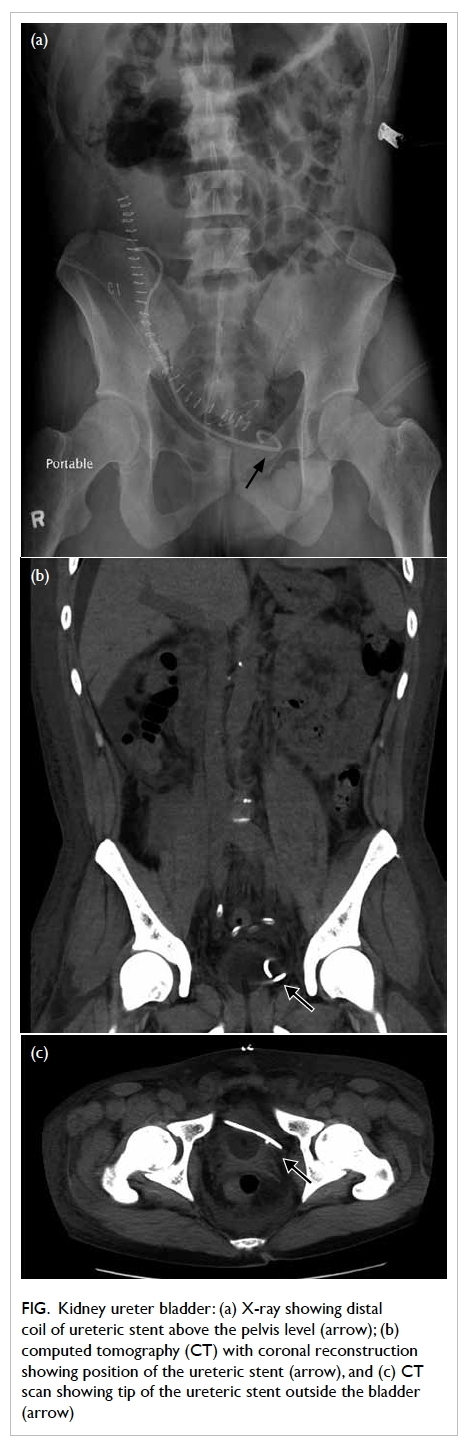
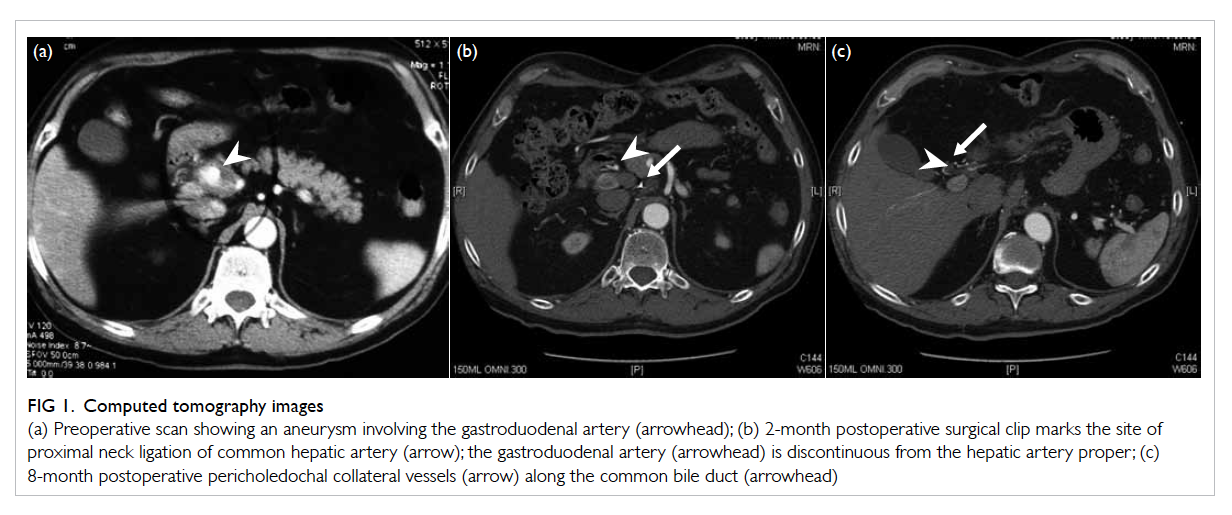
no hydronephrosis. Ultrasound-guided renal biopsy
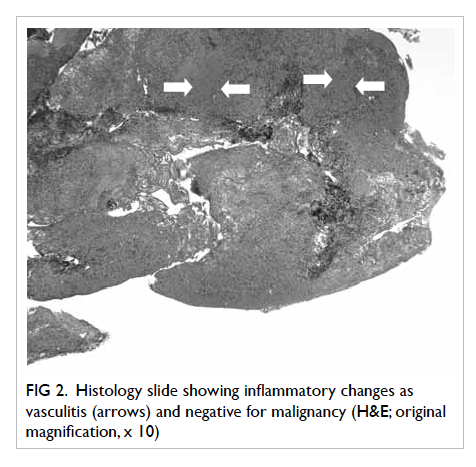
was performed and the histology showed features
of tubulointerstitial nephritis with eosinophil-rich
interstitial infiltrates and occasional granulomas
(Fig). The likely diagnosis was drug-induced acute
interstitial nephritis (AIN). Clozapine was withheld,
fever subsided afterwards and renal function
gradually returned to normal 4 weeks following
cessation of clozapine.
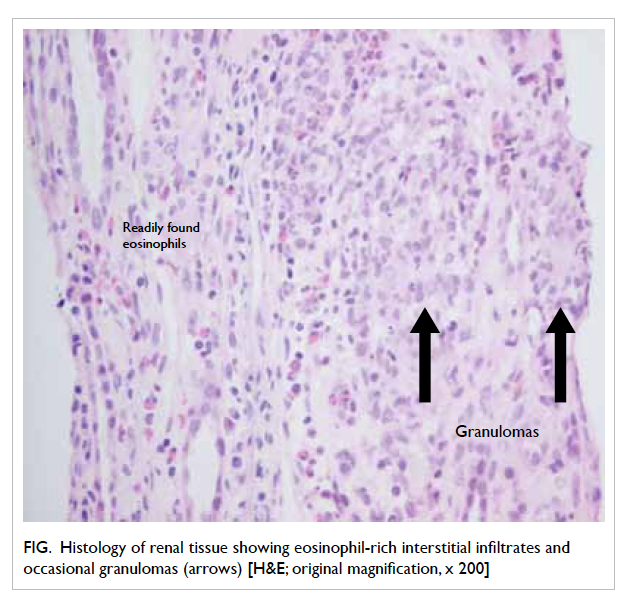
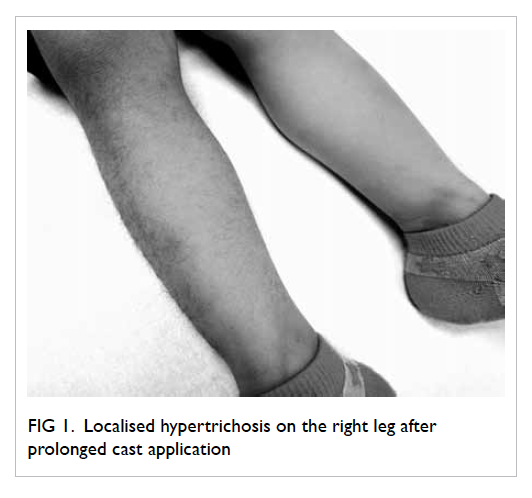
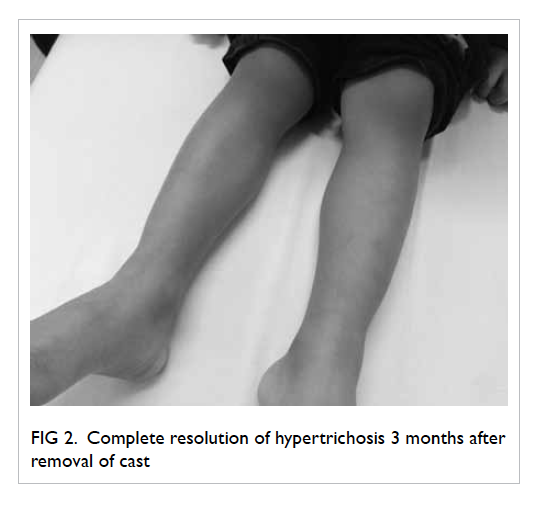
Figure. Histology of renal tissue showing eosinophil-rich interstitial infiltrates and occasional granulomas (arrows) [H&E; original magnification, x 200]
Discussion
Drug-induced AIN is not uncommon. Common
nephrotoxic drugs include non-steroidal anti-inflammatory
drugs (NSAIDs) and antibiotics such
as aminoglycosides and vancomycin. Although
amoxicillin/clavulanic acid remains a possible culprit
in this case for AIN, the short duration of amoxicillin/clavulanic acid use and sequence of events are more
suggestive of clozapine-induced AIN.
Acute interstitial nephritis is defined as an
immune-mediated condition characterised by
the presence of inflammation and oedema in the
tubulointerstitium of the kidneys. It accounts for
15% to 27% of biopsy-proven acute kidney injury.
Acute interstitial nephritis is most commonly
drug-induced although the cause may also be
infective, autoimmune, or idiopathic.1 2 3 4 5 The classic triad—fever, skin rash, and eosinophilia—is only
present in 5% to 10% of patients with AIN.2 3 4 5 As a
result, a high level of clinical suspicion is essential
since around 40% of patients with AIN eventually
required dialysis.2 A detailed drug history, especially
herbal medicine which is common in our locality,
recreational drugs and radio-contrast, are important
for diagnosis. Other history including water
exposure and animal contact (pets or stray animals)
can help exclude or confirm several infective causes
such as Legionnaires’ disease and leptospirosis.
Systemic manifestations of vasculitis should also be
specifically looked for during physical examination.
There are no specific signs and symptoms to
differentiate drug-induced AIN from other causes of
AIN. Fever is present in around 30% of patients with
drug-induced AIN3 5 and it typically occurs within 2 weeks of initiation of a new drug.3 Skin rash, usually
of morbilliform or maculopapular appearance, is
found in only 15% to 50% of patients, depending on
the type of medication.3 5 One retrospective review showed that around 45% of patients have arthralgia,
and 15% and 21% of patients have dysuria and loin
pain, respectively.5 Eosinophilia is thought to have
great diagnostic value in drug-induced AIN as it
signifies a hypersensitivity reaction although the
degree and frequency of eosinophilia vary with
different medications.4 Methicillin-induced AIN is
associated with eosinophilia in 80% of cases while
AIN due to NSAID is less commonly associated
with eosinophilia, which is only around 35% of
cases.4 However, the presence of eosinophilia can
also be found in infections, especially those caused
by helminths, allergic diseases such as asthma
and eczema, and even malignant conditions such
as lymphoma and carcinoma of the colon. Like
eosinophilia, the presence of eosinophiluria is neither
sensitive nor specific for drug-induced AIN. The
sensitivity ranges from 63% to 91%, and specificity
from 52% to 94%.3 4 Other conditions such as lower urinary tract infection, pyelonephritis, prostatitis,
acute tubular necrosis, glomerulonephritis,
and urinary schistosomiasis can also result in
eosinophiluria.3 Other parameters from urine
samples may also provide clues for drug-induced
AIN. Proteinuria is present in 93% of patients but
only 2.5% have a nephrotic range of proteinuria.2
Haematuria is usually microscopic and is present in
60% to 80% of cases while gross haematuria is only
present in 5% of patients.2 Renal biopsy can confirm
the diagnosis of AIN by demonstrating inflammation
and oedema of the renal interstitium and tubulitis.2 3 4 5
The presence of a considerable number of eosinophils
in the interstitium will point to a diagnosis of drug-induced
AIN, while an abundance of neutrophils is
suggestive of an infective cause.3
As drug-induced AIN is idiosyncratic and
not dose-dependent, the mainstay of treatment is
cessation of the causative agents. Nonetheless, not
all patients experience complete recovery despite
withdrawal of the index medication. Neither the
severity of renal failure, the extent of interstitial
involvement (including fibrosis), nor the severity
of tubulitis predicts the clinical outcome.2 3 4 5 More
established prognostic factors are the duration
of renal failure and creatinine level 6 to 8 weeks
after diagnosis.4 Some studies advocate the use
of corticosteroid to hasten recovery and prevent
progression to chronic kidney disease6 but these
studies have usually been small and retrospective.2 3 4
Since 1999, 10 cases of clozapine-induced AIN
have been described.6 7 8 Among these patients, the
ages ranged from 24 to 69 years, and six were male.
In patients who presented within 2 weeks of commencing
clozapine, 80% exhibited a hypersensitivity
reaction. In those who presented late, symptoms
developed up to 3 months after starting clozapine.
Fever was present in 80% but, surprisingly, none
reported skin rash or arthralgia. Proteinuria was
detected in 80% of cases, but only four patients
had red blood cells in the urine. Eosinophilia was
present in half of the patients only. Four patients
had the diagnosis of AIN confirmed by histology.
A phenomenon is observed wherein the effect of
clozapine on the kidney can be potentiated by the
concomitant use of antibiotics, especially those
that are known to have higher risks of interstitial
damage, such as the penicillin derivatives.6 In our
patient, the temporal relationship between the
initiation of clozapine and the development of acute
kidney injury matched the time frame described
in the literature. In addition, the presence of fever,
eosinophilia, and eosinophiluria also raised the
possibility of clozapine-induced AIN, subsequently
confirmed by histology. The renal function of our
patient also improved spontaneously after removing
the index medication.
In conclusion, this case highlights the rare
but important potential side-effect of clozapine.
In addition to monitoring cell counts, regular
monitoring of renal function is recommended
after initiation of clozapine. Early involvement of
nephrologists can provide early recognition of this
entity with prompt investigation and treatment.
References
1. Bellomo R, Kellum JA, Ronco C. Acute kidney injury.
Lancet 2012;380:756-66. Crossref
2. Praga M, González E. Acute interstitial nephritis. Kidney
Int 2010;77:956-61. Crossref
3. Perazella MA, Markowitz GS. Drug-induced acute
interstitial nephritis. Nat Rev Nephrol 2010;6:461-70. Crossref
4. Rossert J. Drug-induced acute interstitial nephritis. Kidney
Int 2001;60:804-17. Crossref
5. Clarkson MR, Giblin L, O’Connell FP, et al. Acute interstitial
nephritis: clinical features and response to corticosteroid
therapy. Nephrol Dial Transplant 2004;19:2778-83. Crossref
6. Kanofsky JD, Woesner ME, Harris AZ, Kelleher JP, Gittens
K, Jerschow E. A case of acute renal failure in a patient
recently treated with clozapine and a review of previously
reported cases. Prim Care Companion CNS Disord
2011;13:PCC.10br01091.
7. An NY, Lee J, Noh JS. A case of clozapine induced acute
renal failure. Psychiatry Investig 2013;10:92-4. Crossref
8. Mohan T, Chua J, Kartika J, Bastiampillai T, Dhillon R.
Clozapine-induced nephritis and monitoring implications.
Aust N Z J Psychiatry 2013;47:586-7. Crossref