Minimally invasive enteroscopically guided small bowel resection
DOI: 10.12809/hkmj144270
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Minimally invasive enteroscopically guided small bowel resection
Tommy CH Man, MB, BS;
KC Ng, MB, BS;
KW Wong, MB, BS;
Francis PT Mok, MB, BS
Department of Surgery, Caritas Medical Centre, Shamshuipo, Hong Kong
Corresponding author: Dr Tommy CH Man (manchunhin@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
Localisation of small bowel pathology is often
difficult, especially intramural small bowel lesions.
Even with the use of laparoscopy, visualisation of
small bowel lesion is not always possible. The most
accurate method to identify such a lesion is by
laparotomy with direct visualisation and palpation
of the lesion. However, the recent trend in surgical
development aims for minimally invasive procedures
while keeping the excision of surgical pathology
safe and complete, with less surgical trauma. This
report illustrates a case of minimally invasive
enteroscopically guided small bowel resection.
Case report
A 64-year-old male with a history of hepatic
carcinosarcoma had his right hemihepatectomy
done in 2011. The resection margins were clear and
interval computed tomography (CT) scans did not
reveal any recurrence.
One year after the surgery he was admitted
for complaints of non-specific abdominal pain,
melena, and dizziness. On admission, the
haemoglobin level was only 50 g/L although his
oesophagogastroduodenoscopy and colonoscopy
were normal. Capsule endoscopy was performed
and this showed a fungating tumour in the jejunum
with evidence of bleeding. Another CT scan was
arranged which confirmed that there was a 2.5-cm
intraluminal lesion in the small bowel, suggesting
a probable occurrence of gastro-intestinal stromal
tumour.
Subsequently, single-port laparoscopic
resection of the small bowel tumour under single-balloon
enteroscopic guidance was performed. The
patient was operated on under general anaesthesia
in supine position with prophylactic antibiotics.
The single-balloon enteroscope (Olympus SIF-Q260;
Olympus Medical Systems Corp, Japan) was
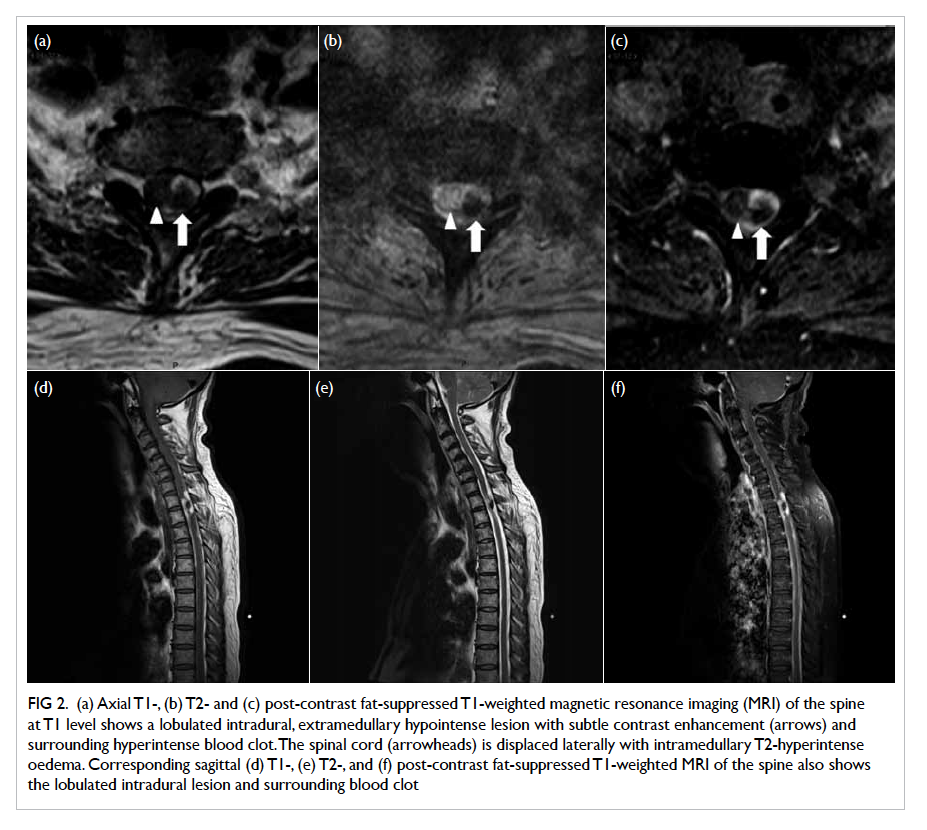
used to locate the site of tumour (Fig 1a). The lesion was marked with endomarker (Fig 1b) and lipiodol
injection, and the enteroscope was left in situ in
order to guide the site of skin incision. Fluoroscopy
and transillumination of the small bowel at the site
of lesion confirmed that the tumour was situated in
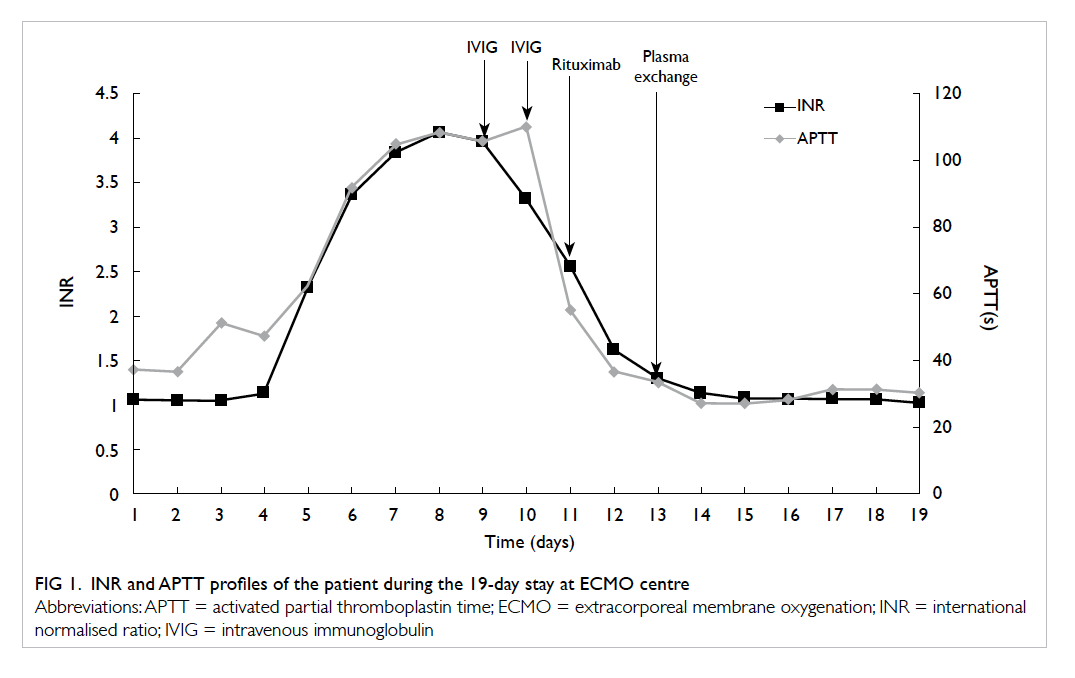
the left upper quadrant of the abdomen (Figs 2a and 2b).
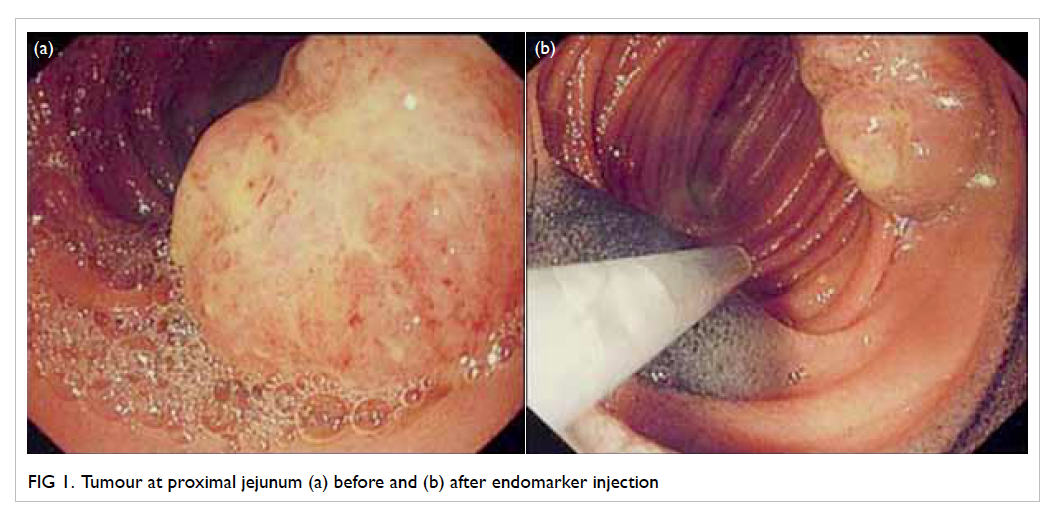
Figure 1. Tumour at proximal jejunum (a) before and (b) after endomarker injection
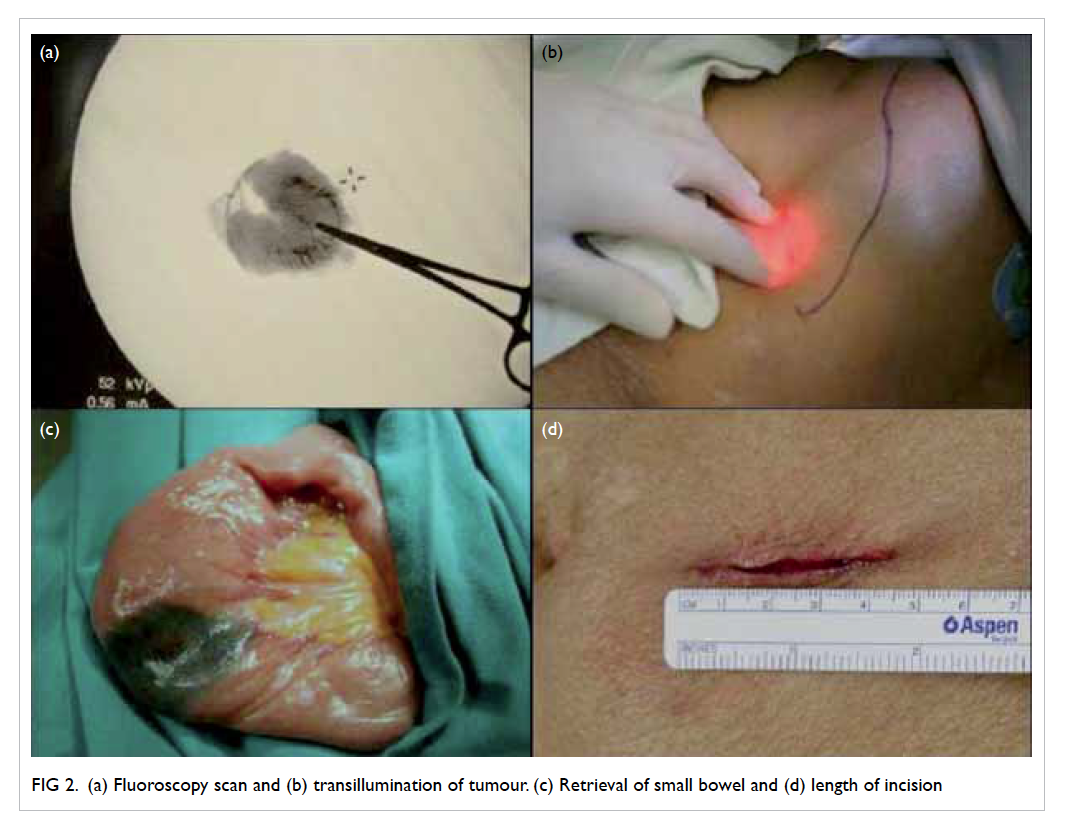
Figure 2. (a) Fluoroscopy scan and (b) transillumination of tumour. (c) Retrieval of small bowel and (d) length of incision
A 2-cm incision was made for laparoscopic
procedure using a Hassan laparoscopic port that was
inserted with an 8-degree laparoscope with working
channel (OLYMPUS A5252A laparoscope; Olympus
Medical Systems Corp, Germany). Laparoscopy
procedure confirmed that there were no suspicious
hepatic and peritoneal nodules. The diseased part
of the small bowel was taken out using a grasper
through the working channel after extension of the
skin wound (Fig 2c). Usual small bowel resection
was done with primary anastomosis using linear
staplers and the entire procedure lasted for about
225 minutes.
The patient tolerated the whole procedure
well and his recovery was satisfactory. He gradually
resumed oral feeding and was fit for discharge on
day 3 after the procedure.
Discussion
Midline laparotomy incision is often a straightforward
procedure for treatment and resection of
small bowel lesions. The long laparotomy wound,
however, may cause a lot of pain, leading to prolonged
hospital stay and more analgesic requirement that
may impair respiratory function especially in patients
with advanced age and multiple co-morbidities.
Although capsule endoscopy for identification
of small bowel pathologies1 2 has become prevalent
in patients with occult gastro-intestinal bleeding,
sometimes it renders the operation difficult to
accurately determine the site of lesion and locate the
tumour.
The recent advances in surgical technology
and development of new techniques have made
minimally invasive procedures possible, like robotic
surgery and endoscopic resection. However, the
installation of these surgical instruments takes up a
lot of space in the operating theatre and increases
the cost of enhancement, not to mention the huge
maintenance cost.3 4 As such, we present a case of
minimally invasive small bowel resection which is
cost-effective to be adopted by most hospitals.
To recap, the aim of minimally invasive surgery
is to reduce surgical morbidity with smaller and less
wound while maintaining pathological clearance
and safety. In our design, the use of enteroscope
provided accurate localisation of the pathology by
direct visualisation.5 After confirming the position
of the pathology, the skin incision could be precisely
made on top of the lesion. Before excising the lesion,
laparoscopy was also performed to make sure that
there was no intra-abdominal metastatic deposit.
The wound was extended to 4.5 cm and this allowed
the diseased small bowel to be retrieved for resection
as in an open procedure (Fig 2d). The instruments
used are readily available in most hospitals and the
procedure can be performed at a relatively lower
cost.
Single-balloon enteroscope, the procedure
used in our case, is a recently developed technology
for diagnosing small bowel pathology. The setup of this safe enteroscope is simple.6 Moreover, some
evidence bore out that the learning curve of single-balloon
enteroscope is insignificant.7 8 Previous
study also showed that single-balloon enteroscope
provided a diagnostic yield similar to double-balloon
enteroscope which requires more technical skills.9
In this operation, three types of localisation
methods were adopted after position of the tumour
was confirmed by enteroscopy. These included
lipiodol injection, transillumination to localise the
incision site, and endomarker injection to find out
precisely where the tumour was located.
Lipiodol injection at the site of tumour allows
localisation of lesion under fluoroscopic guidance,
which is useful to guide the site for skin incision.
It can be done before operation, hence saves time
during intra-operative enteroscopy. The exact
position of the tumour, however, may not be easily
located by laparoscope after identification of the
incision site.
In such situations, injection of endomarker
before operation is recommended during
laparoscopy as this enables better visualisation of
the portion of the diseased bowel to be retrieved
for further resection. However, endomarker cannot
replace lipiodol in localisation of lesion as it is not
radio-opaque and cannot aid localisation of skin
incision site.
Alternatively, transillumination using
enteroscope allows real-time localisation of the
incision site and this is superior to merely injecting
lipiodol. However, the risk of complications arising
from intra-operative enteroscope may be hiked up
and the operating time may be extended.
To facilitate the operation and shorten the
time required, preoperative enteroscopy rather than
intra-operative enteroscopy should be employed.
The injection of lipiodol and endomarker can be
done before operation. As an initial attempt of
our de-novo technique in this case, we decided to use
intra-operative enteroscopy with transillumination
for real-time tumour localisation even though the
operating time was inevitably prolonged.
In addition to the above methods, the use of
endoclip is considered a possible option for accurate
localisation. Since endoclip is radio-opaque, it
can serve both purposes for incision site and
tumour localisation under fluoroscopic guidance.
Nevertheless, the accuracy could be affected due to
the risk of dislodgement of these endoclips.
Conclusion
Enteroscope is a safe option for guiding minimally
invasive small bowel resection with accurate
localisation of pathology. There are different ways
for localisation including the use of endomarker,
lipiodol injection, transillumination as well as
endoclip. Large-scale studies using these techniques
should be considered in order to understand the
efficacy of such newer methods.
References
1. Mavrogenis G, Coumaros D, Bellocq JP, Leroy J. Detection
of a polypoid lesion inside a Meckel’s diverticulum using
wireless capsule endoscopy. Endoscopy 2011;43 Suppl 2
UCTN:E115-6.
2. Mavrogenis G, Coumaros D, Lakhrib N, Renard C, Bellocq JP,
Leroy J. Mixed cavernous hemangioma-lymphangioma
of the jejunum: detection by wireless capsule endoscopy.
Endoscopy 2011;43 Suppl 2 UCTN:E217-8.
3. Amodeo A, Linares Quevedo A, Joseph JV, Belgrano E,
Patel HR. Robotic laparoscopic surgery: cost and training.
Minerva Urol Nefrol 2009;61:121-8.
4. Kim CW, Baik SH. Robotic rectal surgery: what are the
benefits? Minerva Chir 2013;68:457-69.
5. Ress AM, Benacci JC, Sarr MG. Efficacy of intraoperative
enteroscopy in diagnosis and prevention of recurrent
occult gastrointestinal bleeding. Am J Surg 1992;163:94-9. Crossref
6. Yoshiya S, Sugimachi K, Nakamura S, et al. Preoperative
diagnostic value of single-balloon enteroscopy for
successful surgical treatment of three independent-origin
gastrointestinal malignant tumors: report of a case. Surg
Today 2011;41:1007-10. Crossref
7. Upchurch BR, Sanaka MR, Lopez AR, Vargo JJ. The
clinical utility of single-balloon enteroscopy: a single-center
experience of 172 procedures. Gastrointest Endosc
2010;71:1218-23. Crossref
8. Tsujikawa T, Saitoh Y, Andoh A, et al. Novel single-balloon
enteroscopy for diagnosis and treatment of the small
intestine: preliminary experiences. Endoscopy 2008;40:11-5. Crossref
9. Domagk D, Mensink P, Aktas H, et al. Single- vs. double-balloon
enteroscopy in small-bowel diagnostics: a randomized multicenter trial. Endoscopy 2011;43:472-6. Crossref