Three different ophthalmic presentations of juvenile xanthogranuloma
Hong Kong Med J 2014;20:261–3 | Number 3, June 2014
DOI: 10.12809/hkmj134059
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Three different ophthalmic presentations of juvenile xanthogranuloma
Henry HW Lau, FRCS, FHKAM (Ophthalmology)1;
Wilson WK Yip, MB, ChB, FHKAM (Ophthalmology)1;
Allie Lee, MB, BS2;
Connie Lai, MB, BS, FHKAM (Ophthalmology)1;
Dorothy SP Fan, FRCS, FHKAM (Ophthalmology)2
1 Department of Ophthalmology and Visual Sciences, Prince of Wales
Hospital, Shatin, Hong Kong
2 Department of Ophthalmology and Visual Sciences, The Chinese
University of Hong Kong, Hong Kong Eye Hospital, Hong Kong
Corresponding author: Dr Henry HW Lau (henrylau@cuhk.edu.hk)

Abstract
Three cases of juvenile xanthogranuloma from
two ophthalmology departments were reviewed.
Clinical histories, ophthalmic examination, physical
examination, investigations, and treatment of these
cases are described. A 4-month-old boy presented
with spontaneous hyphema and secondary
glaucoma. He was treated with intensive topical
steroid and anti-glaucomatous eye drops. The
hyphema gradually resolved and the intra-ocular
pressure reverted to 11 mm Hg without any other
medication. Biopsy of his scalp mass confirmed the
diagnosis of juvenile xanthogranuloma. A 31-month-old
boy presented with a limbal mass. Excisional
biopsy of the mass was performed and confirmed it
was a juvenile xanthogranuloma. A 20-month-old
boy was regularly followed up for epiblepharon and
astigmatism. He presented to a paediatrician with
a skin nodule over his back. Skin biopsy confirmed
juvenile xanthogranuloma. He had no other ocular signs. Presentation of juvenile xanthogranuloma
can be very different, about which ophthalmologists
should be aware of. Biopsy of the suspected lesion is
essential to confirm the diagnosis.
Introduction
Juvenile xanthogranuloma (JXG) is a benign
histiocytic skin disorder encountered primarily
in infancy and childhood. Approximately 10%
of these patients exhibit ocular manifestations,
whose presentations vary. Although patients can
be asymptomatic, occasionally they have associated
glaucoma and even blindness.1 2
Case reports
Case notes from 1 January 2008 to 31 December 2009
in two ophthalmology departments were reviewed.
Three cases with a diagnosis of JXG were identified.
The clinical histories, ophthalmic examination
findings, physical examination, investigation results,
and treatment of these patients are described.
Case 1
A 4-month-old boy with glucose-6-phosphate
dehydrogenase deficiency was referred by a
paediatrician because of right eye redness for 2
weeks. His antenatal and birth history was otherwise
unremarkable. He had no recent history of eye injury.
On examination, his right eye was diffusely injected,
with a hazy right cornea and irregular pupil. There was a 1-mm organised blood clot in the anterior
chamber of his right eye. No rubeosis was noted.
Left eye examination was unremarkable, with a clear
cornea and no hyphema. Fundal examination of the
left eye was normal. The intra-ocular pressure (IOP)
of right eye was 48 mm Hg and in the left eye it was
14 mm Hg. In both eyes the corneal diameters (10.5
x 10 mm) were normal for his age.
Blood tests—including complete blood
picture, clotting profile, renal and liver function
tests—were performed to rule out metabolic or
haematological abnormalities, but yielded nil
abnormal. Ultrasonography of his right eye showed
a clear vitreous and the retina was flat without any
intra-ocular mass. Magnetic resonance imaging
of the brain and orbits was also performed, but
revealed no intra-ocular mass. X-rays assessing his
bones showed no features of non-accidental injury.
The patient’s medical and family history was non–contributory.
He was treated intensively with 1% topical
prednisolone acetate (Pred Forte ophthalmic
suspension USP, Allegan) 1 drop every hourly and
anti-glaucomatous eye drops including 2% topical
dorzolamide hydrochloride–timolol maleate
ophthalmic solution (Cosopt; MSD) 1 drop twice daily and topical 0.03% bimatoprost ophthalmic
solution (LUMIGAN; Allergan) 1 drop at night. The
hyphema gradually resolved and IOP normalised
(right eye, 14 mm Hg) without medication. No iris
mass was noted after hyphema subsided. There
was a skin nodule over the scalp but no other skin
lesion was identified (Fig a). Biopsy of the scalp
lesion yielded skin tissue with cellular intradermal
expansion by histiocytes, which were uniform with
small vesicular nuclei and foamy cytoplasm. The
features were compatible with JXG.
The hyphema did not recur over the 2-year
follow-up. His IOP remained normal (right eye
11 mm Hg and left eye 15 mm Hg) without any
medication. The latest visual acuity of both eyes was
20/30.
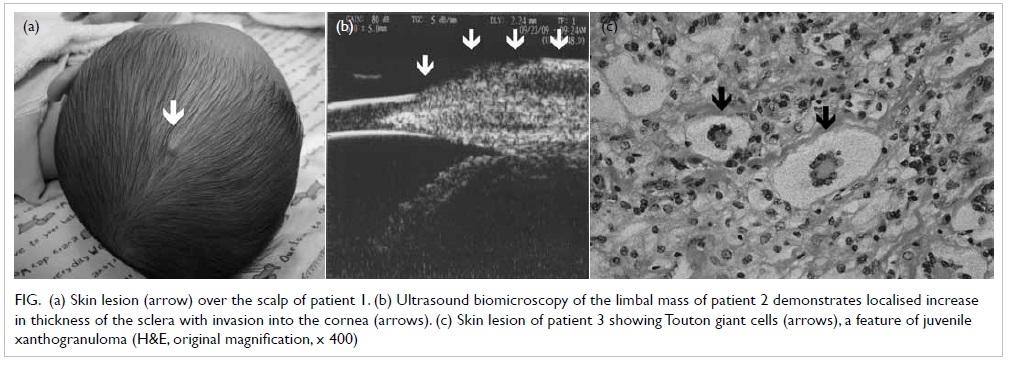
Figure. (a) Skin lesion (arrow) over the scalp of patient 1. (b) Ultrasound biomicroscopy of the limbal mass of patient 2 demonstrates localised increase in thickness of the sclera with invasion into the cornea (arrows). (c) Skin lesion of patient 3 showing Touton giant cells (arrows), a feature of juvenile xanthogranuloma (H&E, original magnification, x 400)
Case 2
The second case was a 31-month-old boy who was born full-term with a normal birth weight (2.7 kg)
and had normal development all along. He presented
to us with an enlarging nasal limbal mass over the
right eye. The mass had been noticed by his mother
for 9 months. The patient was treated elsewhere
with topical steroids and antibiotics but the lesion
was unresponsive. Incisional biopsy of the lesion was
performed and histopathology was reported to show
‘inflammation’. Further increase in size of the lesion
was noted after the biopsy. No other skin lesion was
evident elsewhere. Ultrasound biomicroscopy of
the right eye demonstrated a localised increase in
thickness of the sclera at the site of the lesion with
invasion into the cornea and the borders were ill-defined
(Fig b).
Using Kay Single Pictures, the unaided visual
acuity of the right eye was 20/60 and of the left eye
was 20/40. Examination under general anaesthesia
was performed. There was a right-sided 6-mm limbal
yellowish-grey mass over the nasal region with an
adjacent lipid keratopathy of 1 mm. The other eye
anterior segment examination was unremarkable.
The IOP (right eye 15 mm Hg and left eye 14 mm Hg),
corneal diameter (12.5 mm x 12 mm), and fundal
examination of both eyes were all normal.
The lesion was excised and a partial sclerectomy
was performed. Bared sclera was covered by
conjunctiva. Intra-operatively, the mass did not
show any deep scleral involvement. After removal of
the lesion, the bare area of the sclera was covered
with a conjunctival graft. Histopathology sections
showed a JXG comprising aggregates of histiocytes
and Touton giant cells, situated in a fibrous stroma
covered by non-keratinising squamous epithelium.
Postoperatively he was started on topical 1%
prednisolone acetate ophthalmic suspension USP
(PRED FORTE; Allergan) 1 drop 6 times daily and
topical 0.5% levofloxacin (Cravit; Santen) 1 drop
4 times daily. Recovery was uneventful. Upon
last follow-up 14 months after surgery, the best-corrected visual acuity of both eyes was 20/20 with
no evidence of recurrence.
Case 3
The third case was a 20-month-old boy who was
regularly followed up because of epiblepharon. His
unaided visual acuity of both eyes was 20/50. He also
had astigmatism. Refraction of the right eye was -1.00
D/-1.00 D x 39 and for the left eye it was -1.00 D/
-2.00 D x 157. He presented to a paediatrician with
a skin nodule over his back. Biopsy of the skin lesion
yielded sections with bland epidermis. There was
a well-demarcated nodule in the upper and lower
dermis that was composed of histiocytes and foam
cells, and a few Touton giant cells were seen (Fig c).
These features were compatible with the diagnosis
of JXG. He had no other ocular signs and symptoms
including features of a hyphema or an ocular mass.
Upon last follow-up 15 months after surgery,
unaided visual acuity of both eyes was 20/50 with no
evidence of recurrence.
All three cases yielded a good visual prognosis
and there was no recurrence of the disease.
Discussion
Ocular involvement is the most common
extracutaneous manifestation of JXG. Risk factors
for the development of eye diseases include the
number of skin lesions, and being under 2 years old.3
This condition can affect the orbit, iris, ciliary body,
cornea, and episclera, with the iris being the most
commonly affected.4 Patients can present with iris
nodules which can be quite vascular and may bleed
spontaneously causing hyphema and secondary
glaucoma.5
Zimmerman1 first reported JXG. In his series
of 53 infants and young children with JXG, he
identified five presenting clinical features of intra-ocular
involvement.1 This included an asymptomatic
localised or diffuse iris tumour, unilateral glaucoma,
spontaneous hyphema, red eye with signs of uveitis,
and congenital or acquired iris heterochromia.
However, JXG can sometimes be difficult to be
diagnosed and can mimic melanomas in the eye.6
Ocular JXG can be diagnosed by a skin biopsy if
typical skin lesions are present. However, absence of
skin lesion cannot rule out JXG, because skin lesions
often regress spontaneously. Fifty percent of patients never develop skin lesions and may first present to
the ophthalmologist.7 Treatment depends on the
presenting signs and symptoms. Topical steroids can
be used for hyphema, and anti-glaucomatous eye
drops can be used if there is secondary glaucoma. In
the presence of an ocular mass or skin mass, biopsy
of the suspected lesion is essential to confirming
the diagnosis. Sometimes JXG does not warrant
treatment. However, if extracutaneous involvement
exists, surgery, chemotherapy, or radiotherapy may
become necessary.8
In this case series, the presentation of JXG
was very different in the three patients. Treatment
modalities should be individualised and tailored for
different clinical presentations. Ophthalmologists
should be aware of the various ophthalmic
presentations in JXG. For skin lesions and systemic
signs and symptoms, we should collaborate with
paediatricians and dermatologists to provide holistic
patient care.
Declaration
No conflicts of interest were declared by the authors.
References
1. Zimmerman LE. Ocular lesions of juvenile xanthogranuloma. Nevoxanthoedothelioma. Am J Ophthalmol 1965;60:1011-35.
2. Mocan MC, Bozkurt B, Orhan D, Kuzey G, Irkec M. Juvenile xanthogranuloma of the corneal limbus: report of two cases and review of the literature. Cornea 2008;27:739-42.
3. Chang MW, Frieden IJ, Good W. The risk intraocular juvenile xanthogranuloma: survey of current practices and assessment of risk. J Am Acad Dermatol 1996;34:445-9. CrossRef
4. Chu AC. Juvenile xanthogranuloma. In: Champion RH, Burton JL, Burn DA, Breathnach SM, editors. Rook's textbook of dermatology. 6th ed. Oxford: Blackwell Science; 2004: 2323-5.
5. Vendal Z, Walton D, Chen T. Glaucoma in juvenile xanthogranuloma. Semin Ophthalmol 2006;21:191-4. CrossRef
6. Fontanilla FA, Edward DP, Wong M, Tessler HH, Eagle RC, Goldstein DA. Juvenile xanthogranuloma masquerading as melanoma. J AAPOS 2009;13:515-8. CrossRef
7. Howard J, Crandall A, Zimmerman P, et al. Juvenile xanthogranuloma of the iris of an adult presenting with spontaneous hyphema. Ophthalmic Pract 2001;19:124-9.
8. Hernandez-Martin A, Baselga E, Drolet BA, Esterly NB. Juvenile xanthogranuloma. J Am Acad Dermatol 1997;36:355-67. CrossRef