Hong Kong Med J 2018 Apr;24(2):107–18 | Epub 6 Apr 2018
DOI: 10.12809/hkmj176336
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Outcomes and morbidities of patients who survive
haemoglobin Bart’s hydrops fetalis syndrome: 20-year retrospective review
Wilson YK Chan, FHKAM (Paediatrics)1;
Alex WK Leung, FHKAM (Paediatrics)2; CW Luk, FHKAM
(Paediatrics)3; Rever CH Li, FHKAM (Paediatrics)4;
Alvin SC Ling, FHKAM (Paediatrics)5; SY Ha, FHKAM
(Paediatrics), FHKAM (Pathology)1
1 Department of Paediatrics and
Adolescent Medicine, Queen Mary Hospital, Pokfulam, Hong Kong
2 Department of Paediatrics and
Adolescent Medicine, Prince of Wales Hospital, Shatin, Hong Kong
3 Department of Paediatrics and
Adolescent Medicine, Queen Elizabeth Hospital, Kowloon, Hong Kong
4 Department of Paediatrics and
Adolescent Medicine, Tuen Mun Hospital, Tuen Mun, Hong Kong
5 Department of Paediatrics and
Adolescent Medicine, Princess Margaret Hospital, Laichikok, Hong Kong
Corresponding author: Dr Wilson YK Chan (wilsonykchan@graduate.hku.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Introduction: Haemoglobin Bart’s
hydrops fetalis syndrome was once considered a fatal condition. However,
advances over the past two decades have enabled survival of affected
patients. Data relating to their morbidities and outcomes will help
medical specialists formulate a management plan and parental
counselling.
Methods: All babies with the
syndrome who survived beyond the neonatal period and were subsequently
managed long-term in eight public hospitals in Hong Kong from 1 January
1996 to 31 December 2015 were included. Patient and parent
characteristics, antenatal care, reasons for continuation of pregnancy,
intrauterine interventions, perinatal course, presence of congenital
malformations, stem-cell transplantation details, and long-term
neurodevelopmental outcomes were reviewed.
Results: A total of nine
patients were identified, of whom five were female and four
male. The median follow-up duration was 7 years. All were Chinese and
were homozygous for the Southeast Asian α-thalassaemia deletion. Five of
the nine mothers received antenatal care at a public hospital and opted
to continue the pregnancy after antenatal diagnosis and counselling.
Despite intrauterine transfusions, all babies were born with respiratory
depression and required intubation and mechanical ventilation during the
neonatal period. Hypospadias was identified in all four male infants.
Growth retardation, global developmental delay, and residual
neurological deficits were noted in two-thirds of the patients.
Haematopoietic stem-cell transplantation was performed in two patients,
who became transfusion-independent.
Conclusions: Survival of
patients with Bart’s hydrops fetalis syndrome is possible but not
without short- and long-term complications; local epidemiology is
comparable to that documented for an international registry. Detailed
antenatal counselling of parents with a non-judgemental attitude and
cautious optimism are imperative.
New knowledge added by this study
- This is the first territory-wide multicentre retrospective review of demographic data, morbidities, and outcome of survivors of haemoglobin Bart’s hydrops fetalis syndrome in Hong Kong.
- Intrauterine transfusion is commonly practised in local obstetric units in an attempt to reduce fetal hypoxia and fetal-maternal complications, presumably contributing to survival.
- Prematurity and perinatal respiratory depression are often encountered; intubation, mechanical ventilation, and exchange transfusions are beneficial. Regular hypertransfusion and optimal iron chelation are advocated. Haematopoietic stem-cell transplantation is curative but morbidities and mortalities should not be overlooked.
- Better patient and doctor education is needed, stressing the importance of early accurate diagnosis and the serious sequelae of late presentation.
- Diagnosis should be considered if ultrasonographic features are clinically suggestive, regardless of parents’ mean corpuscular volume, owing to uniparental disomy or non-paternity. Clinical vigilance and prompt specialist referral for ultrasonography and accurate diagnostic testing are crucial to improve maternal-fetal outcomes.
- For parents who opt to continue the pregnancy after diagnosis, meticulous counselling about perinatal and long-term outcomes and morbidities of survivors is imperative.
- Multidisciplinary anticipatory care among obstetricians, pathologists, neonatologists, and haematologists promotes survival, lowers morbidity, and improves long-term outcomes. Patients can now survive beyond childhood, so adult-care physicians can expect to encounter an increasing number of referrals of adult survivors of haemoglobin Bart’s hydrops fetalis syndrome.
Introduction
Haemoglobin Bart’s hydrops fetalis syndrome (BHFS),
also known as homozygous α0-thalassaemia major or homozygous
α-thalassaemia 1, was first described in 1960.1
2 It was considered fatal in the
1960s to 1970s,3 and fetuses often
died in utero, were stillborn or died during the early neonatal period.4 When prenatal screening and
diagnosis for thalassaemia first started in Hong Kong in 1983, BHFS was
advocated as an indication for termination of pregnancy.5 Nonetheless, the availability of intrauterine
transfusions (IUTs)6 7 and intrauterine exchange transfusions (IUETs) enabled
affected fetuses to survive the perinatal period.
Since the world’s first reported case of survival
in 1985 in Canada,8 increasing
numbers of BHFS survivors have been reported worldwide, including in Hong
Kong.9 The traditional view of its
fatality has been challenged,10 11 and in the 1990s there was
lively debate about the ethical concerns surrounding active resuscitation
and treatment of BHFS babies. Regular transfusions and iron chelation
allow BHFS patients to survive even beyond adulthood, and haematopoietic
stem cell transplantation12 13 14 offers a
cure for this disease, albeit at the expense of possible significant
morbidities and compromised quality of life. Long-term morbidities for
this cohort of patients thus become an important issue to address.
Information gathered by this territory-wide retrospective study will
assist physicians in contemplating perinatal management and counselling of
parents.
Methods
Data collection
The setting for this study was all eight public
hospital paediatric haematology units in Hong Kong that care for patients
with transfusion-dependent thalassaemia. Records of patients diagnosed
with BHFS (either antenatally or postnatally) who survived beyond the
neonatal period and who were subsequently managed long-term at those units
from 1 January 1996 to 31 December 2015 were retrieved from the Hong Kong
Hospital Authority’s Clinical Data Analysis and Reporting System using the
International Classification of Diseases diagnostic code 282.7, searching
only for Haemoglobin-Bart’s disease. Data cross-checking was performed
with the help of the Hong Kong Paediatric Haematology and Oncology Study
Group and paediatric haematologists from the eight hospitals.
Information about patient and parent
characteristics, availability of antenatal care and diagnosis, antenatal
ultrasonographic findings, reasons for continuation of pregnancy, use of
intrauterine transfusions, perinatal course, presence of congenital
malformations, subsequent neonatal and long-term neurodevelopmental
outcome, and availability of stem-cell transplantation and subsequent
outcomes were collected and studied. Patients with BHFS who were not born
in Hong Kong were excluded from this study. No missing cases were
identified during the 20-year study period, as confirmed from personal
communications with both paediatric and adult haematologists from all
public hospitals in Hong Kong. As BHFS pregnancies are considered high
risk owing to possible maternal-fetal complications, it was presumed that
no cases would have been managed by private doctors without support from a
neonatal intensive care unit.
Statistical analyses
This study was primarily descriptive in nature.
Statistical analyses were performed with the Statistical Package for the
Social Sciences (SPSS) version 22.0 (IBM Corp., Armonk [NY], United
States). Continuous variables are expressed as median and range.
Ethics approval
This study complied with the Declaration of
Helsinki and approval was obtained from the Institutional Review Board of
The University of Hong Kong / Hong Kong Hospital Authority West Cluster
and all other clusters of participating hospitals. Verbal (parental)
consent was obtained but formal written consent was not required by the
institutional review board, as this retrospective study did not involve
direct patient care (Ref No. HKUCTR-2148).
Results
Demographic and genetic features
Surviving patients with BHFS were identified in
five of the eight local paediatric haematology units. A total of nine
infants were found, of whom five were female and four male. All patients
were Chinese, and all were confirmed to be homozygous for the Southeast
Asian α-thalassaemia deletion. Six pairs of parents were heterozygous for
the Southeast Asian deletion, and three pairs refused genetic testing, one
of which was suspected to be a case of non-paternity (case 7). For case 6,
the mother was heterozygous for the Southeast Asian deletion
(α-thalassaemia 1), whereas the father had an α3.7 single deletion
(α-thalassaemia 2). The child demonstrated maternal uniparental disomy and
isodisomy (Tables 1 and 2).6 13 14 15 16 17 18 19 20
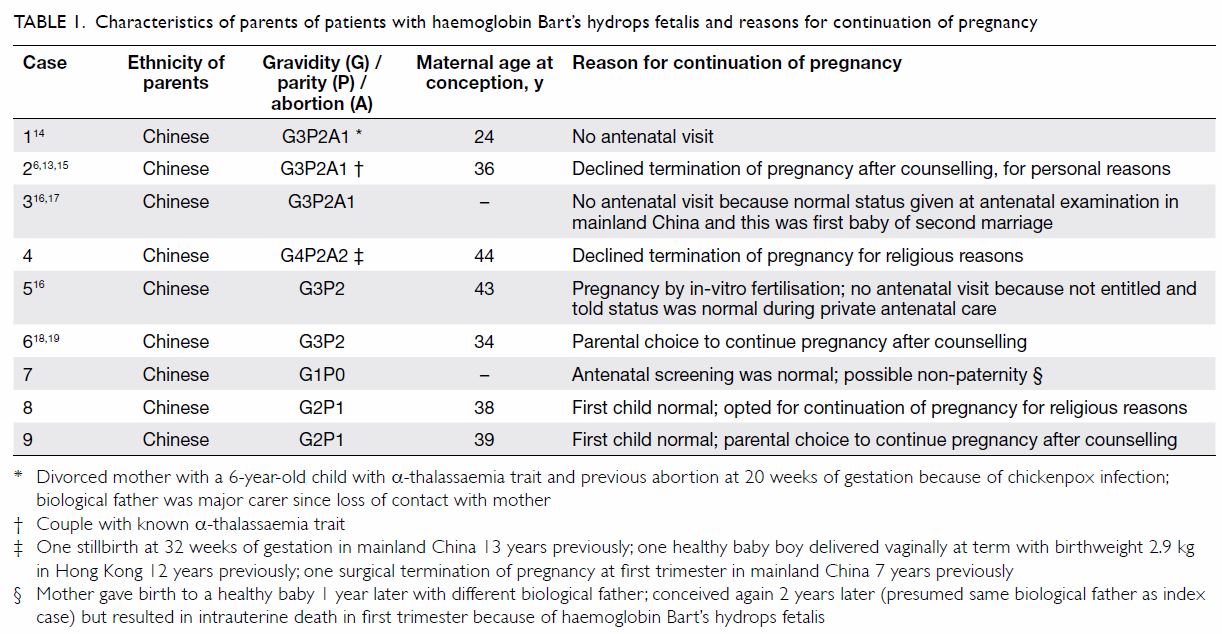
Table 1. Characteristics of parents of patients with haemoglobin Bart’s hydrops fetalis and reasons for continuation of pregnancy
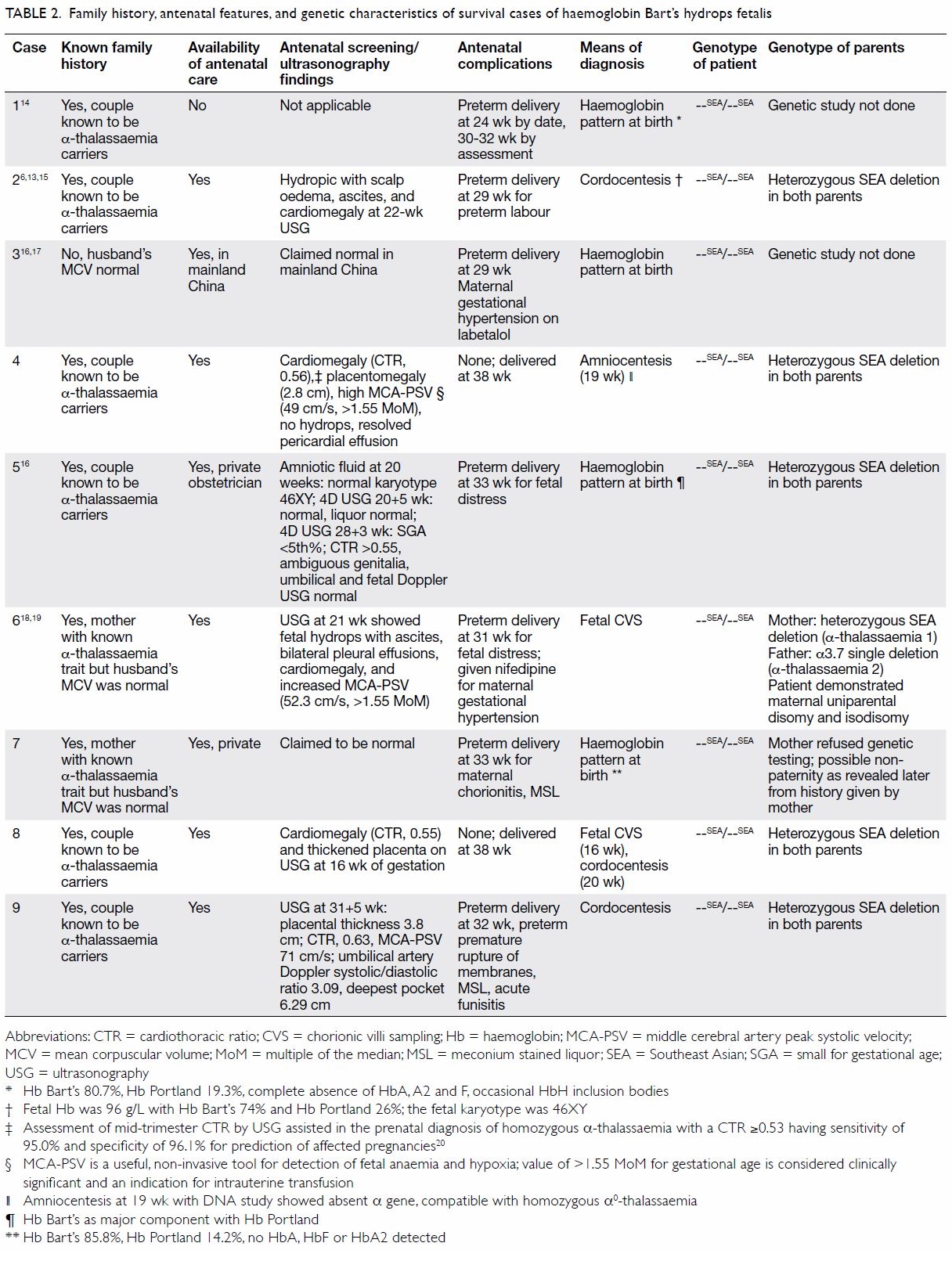
Table 2. Family history, antenatal features, and genetic characteristics of survival cases of haemoglobin Bart’s hydrops fetalis
Prenatal diagnosis, intrauterine management, and
maternal complications
Among the nine mothers, one received no antenatal
care, two received antenatal care in a local private centre, and one
received antenatal care in mainland China. All were considered normal and
received no IUT. For the remaining five mothers who received antenatal
care in a public hospital, all had BHFS diagnosed antenatally in their
neonates (two by cordocentesis, one by chorionic villus sampling, one by
amniocentesis, and one by both cordocentesis and chorionic villus
sampling). All five couples decided to continue the pregnancy after
counselling: two for religious reasons and three out of personal
preference. All five patients had IUT/IUET performed two to four times.
Antenatal ultrasonography of seven fetuses revealed cardiomegaly in four
and hydropic changes in two, one of which subsequently resolved after IUT.
Placentomegaly was detected in three mothers and polyhydramnios in one.
Pre-eclampsia was reported in two mothers and was controlled with
antihypertensive drugs (Tables 2 and 3).6 13 14 15 16 17 18 19 20
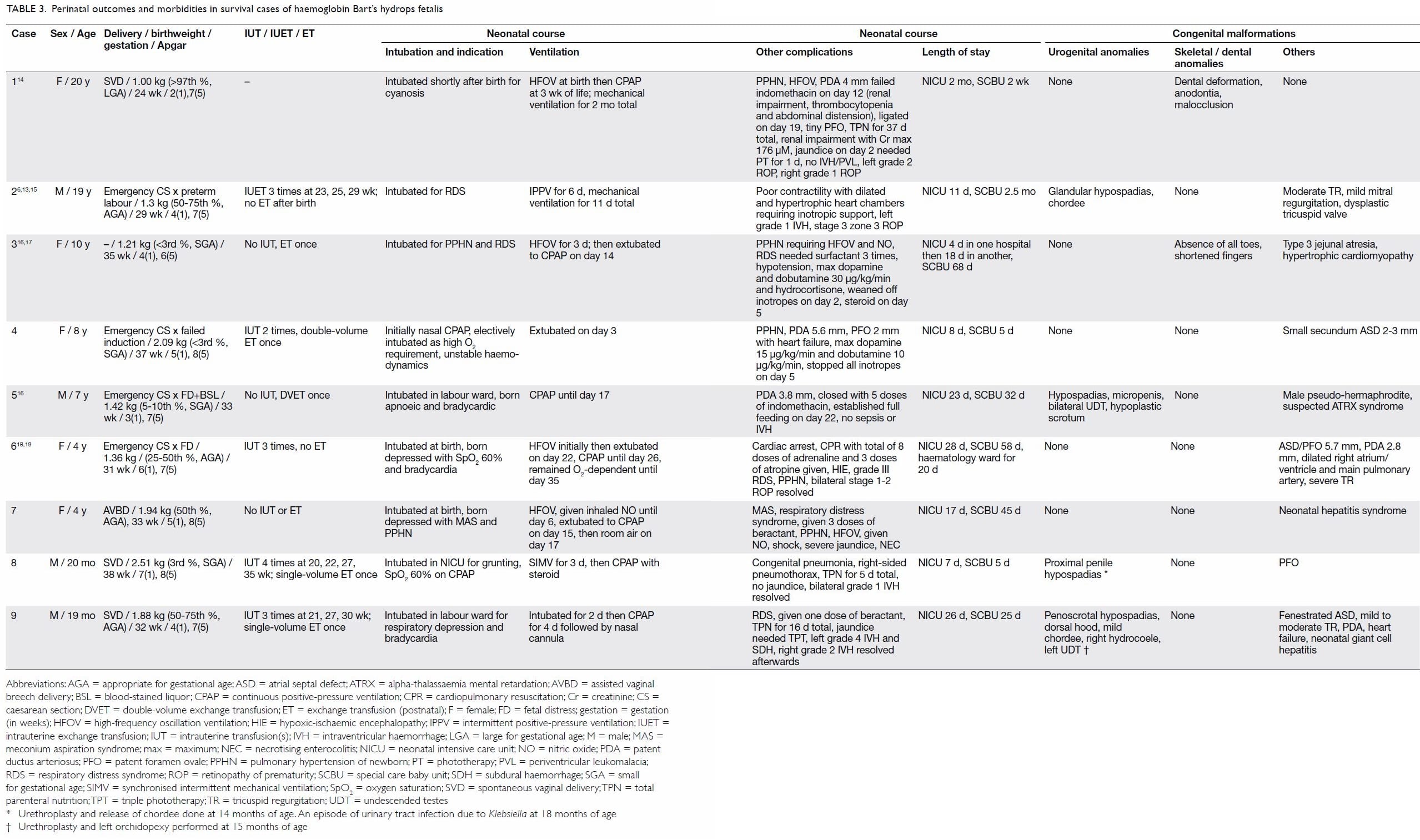
Table 3. Perinatal outcomes and morbidities in survival cases of haemoglobin Bart’s hydrops fetalis
Neonatal outcomes and co-morbidities
Preterm delivery occurred in seven of the nine
cases, with a median gestational age at delivery of 33 weeks. All infants
had respiratory depression at birth and required resuscitation, neonatal
intensive care unit admission, and intubation. Surfactant for respiratory
distress syndrome was required by five infants, and five demonstrated
persistent pulmonary hypertension of the newborn, which required
high-frequency oscillation ventilation and inhaled nitric oxide
administration. Inotropic support with or without hydrocortisone was
required by four infants with haemodynamic instability (poor cardiac
contractility, heart failure, and/or hypotension). In case 6, the infant
required cardiopulmonary resuscitation for more than 20 minutes after
cardiac arrest. Postnatally, exchange transfusion was performed in five
babies: two received a double-volume transfusion and three received a
single-volume transfusion. Three infants received a transfusion within the
first 24 hours of life. The median pre- and post-transfusion haemoglobin
level was 90 and 170 g/L, respectively. All infants showed improved
haemodynamic stability after transfusion. Congenital malformations were
noted in all cases in this cohort (Tables 3 and 4).6 13 14 15 16 17 18 19
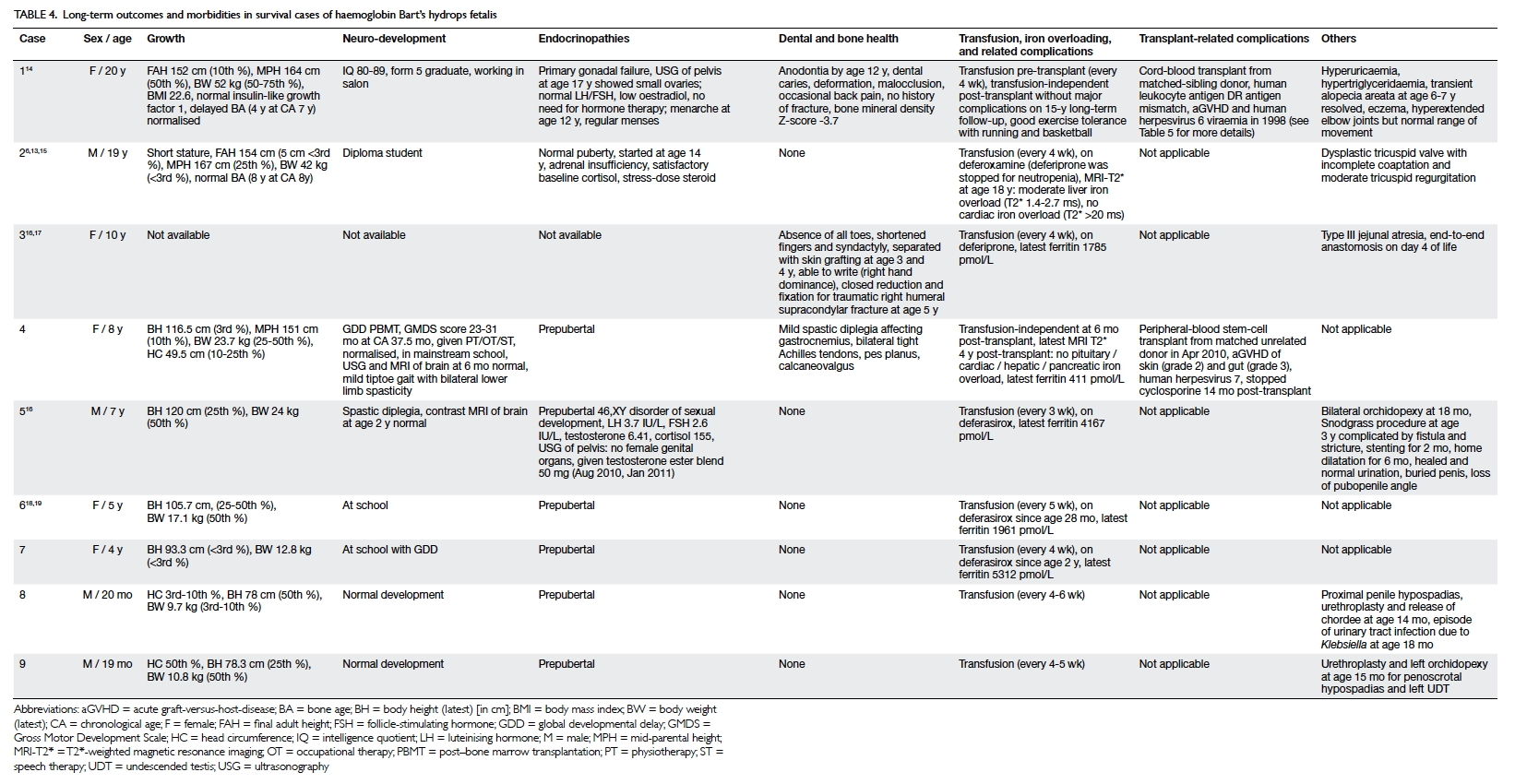
Table 4. Long-term outcomes and morbidities in survival cases of haemoglobin Bart’s hydrops fetalis
All four male babies had hypospadias that required
urethroplasty, and two had concomitant undescended testes that required
orchidopexy. Dental (case 1) and skeletal (case 3) anomalies were noted in
two patients. Regarding the cardiovascular system, patent ductus
arteriosus was noted in five cases and a secundum atrial septal defect in
three. Regarding the digestive system, one infant (case 3) had type 3
jejunal atresia, for which end-to-end anastomosis was performed on day 4
of life. Two patients had neonatal hepatitis: one case resolved with time
and the other still requires regular follow-up for elevated transaminase
levels. No cases of cerebrovascular malformations were identified in this
local cohort.
Growth, puberty, and neurodevelopment
Both survivors who have reached adulthood are of
short stature and have failed to achieve their final adult height, that
is, to reach their predicted mid-parental height. Nonetheless, both had a
normal puberty. Among the nine survivors, two have long-term neurological
deficits, both manifested as mild spastic diplegia, although not affecting
mobility. Five infants had delayed development, one of whom continues to
have borderline low intellect (IQ, 80-89) after reaching adulthood (case
1). Two have normal intellect (cases 2 and 4) despite the need for
multidisciplinary training during infancy, and the remaining two (cases 6
and 7) are attending mainsteam schools that provide extra training and
support. The two cases diagnosed most recently (cases 8 and 9) have had
normal development to date (Tables 3 and 4).6 13 14 15 16 17 18 19
Long-term outcomes and co-morbidities
Two patients received a stem-cell transplant: one
an human leukocyte antigen DR 1 antigen–mismatch sibling-donor cord-blood
transplant, and the other a 10/10 peripheral-blood stem-cell transplant
from a matched unrelated donor. Both patients underwent transplantation at
20 to 21 months of age. Both achieved 100% donor chimerism 1 month after
transplantation and remain transfusion-independent. Of the remaining seven
patients who require regular transfusion every 3 to 6 weeks, only one
shows moderate hepatic iron overload (case 2), and none have demonstrated
infective complications. All five survivors older than 2 years received
iron chelation therapy: three with deferasirox and two with deferiprone
(one of whom has changed to deferoxamine owing to neutropenia). The median
serum ferritin level was 1961 pmol/L (range, 411-5312 pmol/L).
Endocrinopathies were noted in three patients: one had primary gonadal
failure but did not require hormonal replacement therapy (case 1), one had
hypogonadism requiring testosterone (case 5), and one (case 2) had partial
adrenal insufficiency requiring stress-dose steroid but not regular
hydrocortisone replacement (Tables 4 and 5).6 13 14 15 16 17 18 19
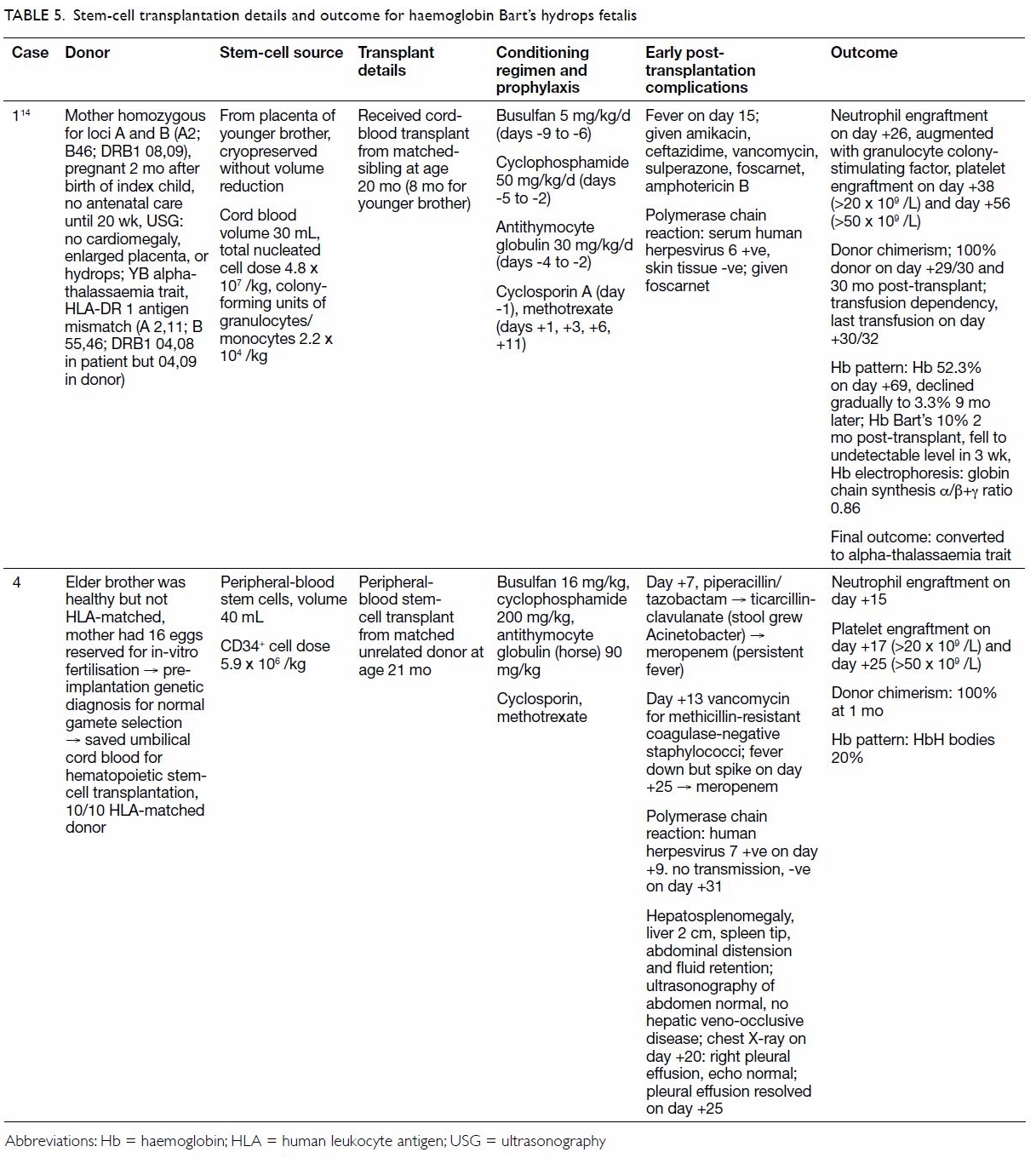
Table 5. Stem-cell transplantation details and outcome for haemoglobin Bart’s hydrops fetalis
Discussion
In Southeast Asia, BHFS is the most common cause of
fetal hydrops.5 21 Because it is an autosomal recessive disorder, when
both parents have two α-globin gene deletions in cis on chromosome 16
(each parent, --/αα), any offspring will have a 25% chance of having BHFS.
In BHFS, haemoglobin tetramers of only gamma chains (γ4) is
ineffective in erythropoiesis and oxygen delivery to tissues. The ensuing
anaemia and tissue hypoxia interfere with organogenesis and development
and also lead to fetal heart failure, extramedullary erythropoiesis, and
maternal complications. In contrast to --FIL and --THAI
gene deletions reported in the Philippines and Thailand, respectively, the
--SEA or Southeast Asian deletion (the most common mutation in
Hong Kong and demonstrated in all nine BHFS cases in this study) affects
only the α-globin gene while sparing the embryonic ζ-globin gene, thus
permitting production of Portland 1 (ζ2γ2) and
Portland 2 (ζ2β2) haemoglobins. Hence, the affected
fetus can survive through the antenatal and early neonatal period.
Pitfalls in prenatal screening and diagnosis
In Hong Kong, prenatal screening using a cut-off
for maternal mean corpuscular volume of ≤80 fL and prenatal diagnosis
using chorionic villus sampling, amniocentesis, and cordocentesis have
been practised since 1983,22 23 24 25 thereby contributing to a decline in BHFS incidence
for two decades. Despite public health endeavours in prenatal screening,
however, BHFS babies continue to be born without prior prenatal diagnosis
or parental counselling, resulting in adverse maternal and fetal outcomes.26 Causes of this phenomenon are
principally two-fold: lack of proper antenatal screening and diagnosis, as
well as improper implementation of screening or diagnostic procedures (Table 1). Better public education in both mainland
China and Hong Kong would rectify the situation. Such education should
stress the importance of early accurate prenatal diagnosis and the
possible serious sequelae of late presentation or delayed diagnosis.
Obstetricians should also note that normal paternal mean corpuscular
volume does not exclude fetal BHFS because of the rare occurrence of
maternal uniparental disomy (case 6)27
and non-paternity (possible in case 7).27
Routine mid-trimester scanning is imperative and diagnosis of BHFS should
be considered if ultrasonography or clinical features are suggestive of
BHFS (cardiomegaly, placentomegaly,18
28 and hydrops), regardless of the
parents’ mean corpuscular volume.27
Placental thickness measurement allows early detection of BHFS in the
first trimester, even before the appearance of hydropic features.18 28 If
hydropic changes are detected, confirmation by cordocentesis and
haemoglobin electrophoresis is warranted.
Counselling for parents in Hong Kong who opt to
continue pregnancy
Suggested salient points of counselling for parents
who opt for continuation of pregnancy are shown in the online
supplementary Appendix. Once considered fatal, BHFS can now be
detected and diagnosed antenatally, with survival being possible albeit
not without complications. Detailed antenatal counselling for parents who
are contemplating continuation of an affected pregnancy is crucial.
Possible maternal-fetal complications, such as gestational hypertensive
disorder and intrauterine growth restriction or death, should be
addressed. On the basis of local experience, IUT is advised because there
is a risk of miscarriage or intrauterine infection (case 9). Multiple IUTs
may be indicated if fetal anaemia is suggested by serial Doppler
ultrasonography (peak systolic velocity of the middle cerebral artery of
>1.5 multiples of the median). Premature delivery and perinatal
respiratory depression are often encountered. Neonatal intensive care unit
admission and intubation are anticipated from local experience. Inotropic
support may be required in the early neonatal period, as well as
cardiopulmonary support, such as mechanical ventilation, surfactant
treatment, high frequency oscillation ventilation, and nitric oxide
inhalation for persistent pulmonary hypertension of the newborn. Exchange
transfusion is often performed postnatally, in most cases within the first
24 hours of life.
Congenital malformations are often encountered, the
most common being genitourinary and musculoskeletal defects, but are
usually amenable to surgical correction. It is worth noting that all male
babies in our local cohort displayed hypospadias. Two-thirds of our
patient cohort showed growth retardation, global developmental delay,
and/or long-term residual neurological deficits. These findings are
comparable to those from an international case series29 and an overseas case report.11
Lifelong hypertransfusions every 4 to 6 weeks and iron chelation are
expected for BHFS survivors. Haematopoietic stem-cell transplantation is a
possible cure and has been successful in some local cases (Table
3). Nevertheless, transplant-related mortality and morbidity should
not be overlooked. The proposal that parents produce a subsequent sibling
to serve as a ‘saviour baby’ and potential donor is feasible but raises
ethical concerns. Careful consideration and proper parental counselling
are necessary.
Comparison of outcomes and morbidities between local
and international cohorts
Globally, 69 survivors are reported in the
international BHFS registry29 with
our local cohort (n = 9) contributing about one-seventh of cases.
Approximately two-thirds of all cases used IUT29
(41/69; 59%), which is similar to the proportion of local cases (5/9;
56%). Globally29 and locally, most
infants were delivered prematurely (respectively, 47/66; 71% and 7/9;
78%). Approximately one-fifth (14/69) of all BHFS survivors underwent
stem-cell transplantation,29 again
similar to our local situation (2/9). Congenital anomalies were present in
all of the local patients, compared with two-thirds (37/55; 64%)
worldwide,29 although urogenital
and limb defects remained the most common. Nearly half (26/55; 47%) of
global BHFS survivors demonstrated various degrees of transient or
permanent neurodevelopmental impairment,29
in contrast to two-thirds of our cohort. Sohan et al (2002)30 described the first BHFS survivor in the United
Kingdom: a 38-week-old baby girl of Hong Kong parents, who was referred at
21 weeks of gestation for hydrops fetalis, received serial IUT and had
BHFS antenatally diagnosed. Two exchange transfusions were performed
postnatally and the baby was discharged on day 6 of life followed by
transfusions every 4 to 5 weeks. At the time of that publication, she was
18 months old and had normal growth and development apart from bilateral
transverse palmar creases and a mild lobster-claw deformity of the right
foot.30
Strengths, limitations, and recommendations
This is the first territory-wide multicentre
retrospective study to describe in depth the basic demographic
characteristics and perinatal and long-term outcomes of BHFS survivors in
Hong Kong over the past two decades or so. It also explores the reasons
and cultural circumstances for which parents opted to continue the
pregnancy despite public health endeavours to promote antenatal screening.
Furthermore, it adds two more local cases to those submitted and recently
published by the BHFS International Consortium.29
However, the small sample size precludes statistical analyses, and the
data covers only the eight local public hospitals with haematology units
and a period of 20 years (as the cut-off age in Hong Kong for paediatric
care is 20 years and the Clinical Data Analysis and Reporting System began
only in 1996). Survivors beyond 20 years of age and patients who defaulted
follow-up to receive long-term medical care in the private sector or
overseas were excluded from this study. In addition, the numbers of
abortions and stillbirths, as well as BHFS babies with early neonatal
death were not studied. Multidisciplinary collaboration between
obstetricians, paediatric haematologists, and adult-care physicians at all
local hospitals and concerted efforts in data collection and analysis are
recommended. With the establishment of the Hong Kong Children’s Hospital
in 2018, it is hoped that a standardised protocol of management and
counselling can be compiled, data collection streamlined, and analysis
facilitated for future research.
To conclude, survival of patients with BHFS is
possible but not without short- and long-term complications. Local
epidemiology of BHFS survivors is similar to that reported for an
international registry. Detailed antenatal counselling of parents with a
non-judgemental attitude and cautious optimism are imperative.
Supplementary information
Online supplementary information (Appendix)
is available for this article at www.hkmj.org.
Acknowledgements
We thank the Hong Kong Paediatric Haematology and
Oncology Study Group and the paediatric haematologists from the eight
participating hospitals for patient management, case contribution, and
data cross-checking.
Declaration
The authors have no conflicts of interest to
disclose.
References
1. Lie-Injo LE, Hie JB. Hydrops foetalis
with a fast-moving haemoglobin. Br Med J 1960;2:1649-50. Crossref
2. Lie-Injo LE. Alpha-chain thalassemia and
hydrops fetalis in Malaya: report of five cases. Blood 1962;20:581-90.
3. Weatherall DJ, Clegg JB, Boon WH. The
haemoglobin constitution of infants with the haemoglobin Bart’s hydrops
foetalis syndrome. Br J Haematol 1970;18:357-67. Crossref
4. Boon WH. Further studies in Bart’s
hydrops foetalis in Singapore. J Singapore Paediatr Soc 1970;12:79-84.
5. Liang ST, Wong VC, So WW, Ma HK, Chan V,
Todd D. Homozygous alpha-thalassaemia: clinical presentation, diagnosis
and management. A review of 46 cases. Br J Obstet Gynaecol 1985;92:680-4.
Crossref
6. Fung TY, Lau TK, Tam WH, Li CK. In utero
exchange transfusion in homozygous alpha-thalassaemia: a case report.
Prenat Diagn 1998;18:838-41.
7. Lee SY, Chow CB, Li CK, Chiu MC. Outcome
of intensive care of homozygous alpha-thalassaemia without prior
intra-uterine therapy. J Paediatr Child Health 2007;43:546-50. Crossref
8. Beaudry MA, Ferguson DJ, Pearse K,
Yanofsky RA, Rubin EM, Kan YW. Survival of a hydropic infant with
homozygous alpha-thalassemia-1. J Pediatr 1986;108(5 Pt 1):713-6. Crossref
9. Lam TK, Chanh V, Fok TF, Li CK, Feng CS.
Long-term survival of a baby with homozygous alpha-thalassemia-1. Acta
Haematol 1992;88:198-200. Crossref
10. Thornley I, Lehmann L, Ferguson WS,
Davis I, Forman EN, Guinan EC. Homozygous alpha-thalassemia treated with
intrauterine transfusions and postnatal hematopoietic stem cell
transplantation. Bone Marrow Transplant 2003;32:341-2. Crossref
11. Lücke T, Pfister S, Dürken M.
Neurodevelopmental outcome and haematological course of a long-time
survivor with homozygous alpha-thalassaemia: case report and review of the
literature. Acta Paediatr 2005;94:1330-3. Crossref
12. Chik KW, Shing MM, Li CK, et al.
Treatment of hemoglobin Bart’s hydrops with bone marrow transplantation. J
Pediatr 1998;132:1039-42. Crossref
13. Ng PC, Fok TF, Lee CH, et al. Is
homozygous alpha-thalassaemia a lethal condition in the 1990s? Acta
Paediatr 1998;87:1197-9. Crossref
14. Zhou X, Ha SY, Chan GC, et al.
Successful mismatched sibling cord blood transplant in Hb Bart’s disease.
Bone Marrow Transplant 2001;28:105-7. Crossref
15. Fung TY, Kin LT, Kong LC, Keung LC.
Homozygous alpha-thalassemia associated with hypospadias in three
survivors. Am J Med Genet 1999;82:225-7. Crossref
16. Kwan WY, So CH, Chan WP, Leung WC,
Chow KM. Re-emergence of late presentations of fetal haemoglobin Bart’s
disease in Hong Kong. Hong Kong Med J 2011;17:434-40.
17. Lee SY, Li CK, Ling SC, Shiu YK.
Survival of homozygous alpha-thalassemia with aplasia/hypoplasia of
phalanges and jejunal atresia. J Matern Fetal Neonatal Med 2009;22:711-3.
Crossref
18. Kou KO, Lee H, Lau B, et al. Two
unusual cases of haemoglobin Bart’s hydrops fetalis due to uniparental
disomy or non-paternity. Fetal Diagn Ther 2014;35:306-8. Crossref
19. Au PK, Kan AS, Tang MH, et al. A fetus
with Hb Bart’s disease due to maternal uniparental disomy for chromosome
16. Hemoglobin 2016;40:66-9. Crossref
20. Tongsong T, Wanapirak C,
Sirichotiyakul S, Chanprapaph P. Sonographic markers of hemoglobin Bart
disease at midpregnancy. J Ultrasound Med 2004;23:49-55. Crossref
21. Wasi P, Na-Nakorn S, Pootrakul S. The
alpha-thalassaemias. Clin Haematol 1974;3:383-410.
22. Chan TK, Chan V, Todd D, Chosh A, Wong
LC, Ma HK. Prenatal diagnosis of alpha-and beta-thalassemias: experience
in Hong Kong. Hemoglobin 1988;12:787-94. Crossref
23. Rubin EM, Kan YW. A simple sensitive
prenatal test for hydrops fetalis caused by alpha-thalassaemia. Lancet
1985;325(8420):75-7. Crossref
24. Kan YW, Bellevue R, Rieder RF, Valenti
C, Dozy AM. Prenatal diagnosis of a-thalassemia. Clin Res 1974;22:374A.
25. Kan YW, Golbus MS, Dozy AM. Prenatal
diagnosis of alpha-thalassemia. Clinical application of molecular
hybridization. N Engl J Med 1976;295:1165-7. Crossref
26. Lee AC, Wong KW, So KT, Cheng MY. Why
are thalassaemia patients born when prenatal screening is available? Hong
Kong Med J 1998;4:121-4.
27. Dame C, Albers N, Hasan C, et al.
Homozygous alpha-thalassaemia and hypospadias—common aetiology or
incidental association? Long-term survival of Hb Bart’s hydrops syndrome
leads to new aspects for counselling of alpha-thalassaemic traits. Eur J
Pediatr 1999;158:217-20. Crossref
28. Ghosh A, Tang MH, Lam YH, Fung E, Chan
V. Ultrasound measurement of placental thickness to detect pregnancies
affected by homozygous alpha-thalassaemia-1. Lancet 1994;344:988-9. Crossref
29. Songdej D, Babbs C, Higgs DR, BHFS
International Consortium. An international registry of survivors with Hb
Bart’s hydrops fetalis syndrome. Blood 2017;129:1251-9. Crossref
30. Sohan K, Billington M, Pamphilon D,
Goulden N, Kyle P. Normal growth and development following in utero
diagnosis and treatment of homozygous alpha-thalassaemia. BJOG
2002;109:1308-10. Crossref