Hong Kong Med J 2014 Dec;20(6):481–5 | Epub 18 Jul 2014
DOI: 10.12809/hkmj144227
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Reconstructive surgery for females with
congenital adrenal hyperplasia due to
21-hydroxylase deficiency: a review from the
Prince of Wales Hospital
CH Houben, MD, FRCSEd; SY Tsui, MB, ChB, MRCS; JW Mou, MB, ChB, FHKAM (Surgery); KW Chan, MB, ChB, FHKAM (Surgery); YH Tam, MB, ChB, FHKAM (Surgery); KH Lee, MB, BS, FHKAM (Surgery)
Division of Paediatric Surgery and Paediatric Urology, Department of Surgery, Prince of Wales Hospital, Shatin, Hong Kong
Part of this research was presented at the RCSEd / CSHK Conjoint
Scientific Congress 2012 (Poster), Hong Kong, 22-23 September 2012
Corresponding author: Dr CH Houben (chhouben@web.de)
 Full
paper in PDF
Full
paper in PDF
Abstract
Objectives: To present the results of feminising
genitoplasty done in female patients with congenital
adrenal hyperplasia due to 21-hydroxylase deficiency.
Design: Case series.
Setting: A tertiary referral centre in Hong Kong.
Patients: Female patients with congenital adrenal
hyperplasia undergoing corrective surgery for
virilisation between 1993 and 2012.
Main outcome measures: The operative result
was judged with a scoring system (1-3) for four
areas: appearance of clitoris, labia and vagina, plus
requirement for revision surgery.
Results: A total of 23 female patients with congenital
adrenal hyperplasia with a median age of 17.5 (range,
1.5-33.8) years were identified. Of these individuals,
17 presented in the neonatal period and early infancy,
of which four had an additional salt-losing crisis. Six
patients—including four migrants from mainland
China—were late presenters at a median age of
2 (range, 0.5-14) years. Twenty-two patients had
corrective surgery at a median age of 2 (range, 1-14)
years. Clitoral reduction was performed in all, and
further surgery in 21 patients. The additional surgery
was flap vaginoplasty in 10 patients, a modified
Passerini procedure in six, and a labial reconstruction
in five; one patient with prominent clitoris was
for observation only. Minor revision surgery (eg
mucosal trimming) was required in three patients;
a revision vaginoplasty was done in one individual.
Of the 23 patients, 18 (78%) with a median age of 20
(range, 9.3-33.8) years participated in the outcome
evaluation: a ‘good’ outcome (4 points) was seen in
12 patients and a ‘satisfactory’ (5-9 points) result in
five patients.
Conclusions: Nearly three quarters of our cohort
(n=17) presented with classic virilising form of
21-hydroxylase deficiency. Only four (25%) patients
experienced a salt-losing crisis. Female gender
assignment at birth was maintained for all individuals
in this group. ‘Good’ and ‘satisfactory’ outcomes of
surgery were reported in nearly all participants.
New knowledge added by this
study
- This is the most comprehensive analysis of the surgical management of congenital adrenal hyperplasia (CAH) in Asian women.
- CAH presenting as salt-losing crisis was seen in less than 25% of this cohort.
- In our region, a proportion of young women (eg migrants) may present late for corrective surgery.
- Early gender assignment in conjunction with the primary carers (parents) and the multidisciplinary team is the preferred option in this Asian community.
- The first 2 years of life is the preferred time for reconstructive surgery in this condition. Notwithstanding, some women may present late in our region.
Introduction
Congenital adrenal hyperplasia (CAH) is a group
of autosomal recessive disorders representing, by
far, the most common disorder of sex development for XX karyotype.1 2 The condition is characterised
by impaired cortisol synthesis; the most frequent
defect—present in approximately 95% of the
patients—is 21-hydroxylase deficiency. The enzyme deficiency leads to a block in cortisol synthesis
followed by a build-up of cortisol precursors
which, in turn, are diverted to androgen synthesis.1
The increased androgen concentration triggers a
variable degree of virilisation in female newborns.
Approximately 75% of patients with this classic
form of CAH have an additional mineralocorticoid
deficiency leading to salt-wasting, failure to thrive
or even hypovolaemic shock.1 Mild forms of CAH
present, typically, later in life with a variable degree
of increased androgen excess.
We present the outcome of a cohort of Asian
individuals with genital ambiguity secondary to
21-hydroxylase deficiency. The emphasis is on
presentation, surgery performed, and the anatomical
and cosmetic outcomes.
Methods
This was a retrospective review of demographics
and presentation of patients with CAH secondary to
21-hydroxylase deficiency who were managed by a multidisciplinary team at our tertiary referral centre
between 1993 and 2012.
Generally, surgery is considered at the age of 12
to 24 months as a one-stage feminising genitoplasty;
clitoral reduction plus further corrective surgery
is performed depending on the intra-operative
findings. This could range from a labiaplasty to flap
vaginoplasty or modified Passerini procedure.3 4 5
Following the initial healing phase, dilatation
of the vagina is recommended according to our
protocol until the tissue is supple, usually after a
few months. At the time of puberty, the vagina is
again assessed and dilatations are recommended, as
necessary, by the gynaecologist. Highly motivated
patients achieve an appropriate-sized vagina with
daily dilatations within a couple of months or even
weeks.
The operative treatment was planned as a one-stage
procedure at our institution. The results of this
cohort were assessed as part of the ongoing review in
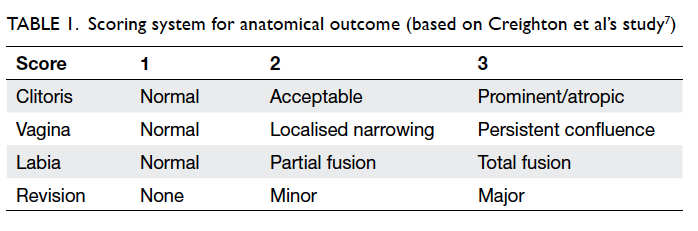
the out-patient setting in 2012/13. A scoring system
(1-3) established previously was used for four areas:
appearance of the clitoris, labia and vagina, plus
requirement for revision surgery.6 7
The criteria for scoring each were as follows:
1—designated for a near-normal appearance, 2—only a medically trained person would be able to
see the result of an intervention, and 3—other
appearances.6 7 The necessity for revision surgery
constituted the fourth factor in the evaluation: score
of 1 for no revision, 2 for minor revision, and 3 for
a major revision. Overall marks of 4 points were
classified as ‘good’, marks of 5 to 9 points ‘satisfactory’,
and marks from 10 to 12 points ‘unsatisfactory’
(Table 1).
Results
Twenty-three females with 21-hydroxylase
deficiency and a median age of 17.5 (range, 1.5-33.8)
years timed on 31 December 2012 were identified.
Seventeen patients presented in the
neonatal period and early infancy, of which four
had an additional salt-losing crisis. Six patients
were late presenters at a median age of 2 (range,
0.5-14) years. Four of these were migrants from
mainland China diagnosed at the age of 1, 3, 8,
and 14 years, respectively. The diagnosis was
established by identifying increased levels of
17-hydroxyprogesterone.
The sex of rearing in the group of neonates
was decided in consultation between the parents,
paediatric endocrinologists, and paediatric
urologists. All patients, including the late presenters,
persisted with the female gender assigned at birth.
Twenty-two patients had corrective surgery at a
median age of 2 (range, 1-14) years. Clitoral reduction
was performed in all 22, and further surgery in 21
patients. The additional surgery consisted of flap vaginoplasty in 10 patients, modified Passerini
procedure in six, and labial reconstruction in five.
One patient with prominent clitoris and otherwise
normal appearance of the vaginal and urethral
opening is currently for observation only (Fig).
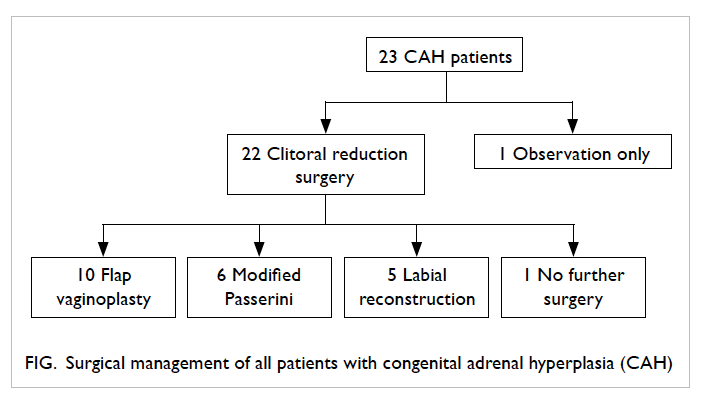
Figure. Surgical management of all patients with congenital adrenal hyperplasia (CAH)
Eighteen individuals with a median age of 20
(range, 9.3-33.8) years were part of the outcome
evaluation in 2012/13. The other five patients
below the age of 5 years were considered too
young for a meaningful assessment. At the time of
diagnosing CAH, all 18 patients had an enlarged
clitoris; separate openings for vagina and urethra
were seen in six individuals, low (intrasphincteric)
confluence in six, and high (suprasphincteric)
confluence in other six individuals. Table 2
summarises the results in accordance with the
initial anatomical findings—high confluence,
low confluence, and separate openings for vagina
and urethra. Minor revision surgery (eg mucosal
trimming) was required in three patients. A ‘good’
outcome was seen in 12 patients and ‘satisfactory’
result in five; one required a revision vaginoplasty
(Table 2).
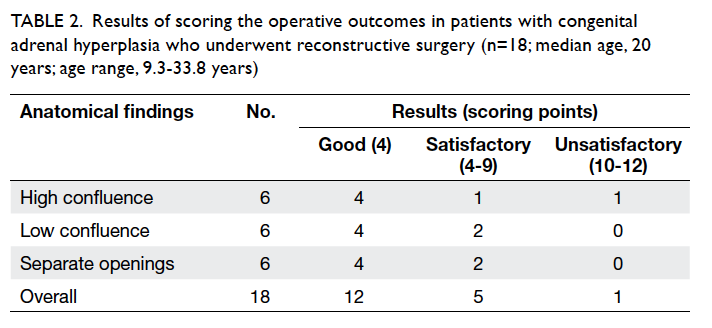
Table 2. Results of scoring the operative outcomes in patients with congenital adrenal hyperplasia who underwent reconstructive surgery (n=18; median age, 20 years; age range, 9.3-33.8 years)
On follow-up, it was noted that all patients
older than 12 years (n=15) experienced menarche
without any obstruction of menstrual blood flow;
13 individuals had regular cycles; one had her cycle
supported by medication, and one patient started
menstruating in the last 6 months, but her cycles
remained irregular.
Two women have given birth by caesarean
section. Both women have two healthy children each,
following successive pregnancies. They delivered
successfully by caesarean section as recommended
by the multidisciplinary team.8 A third woman is
currently pregnant.
A systematic interview including a
psychological evaluation was not part of this review,
but a tendency to more masculine behaviour traits
within this cohort of women was a persistent
observation by clinical staff.
Discussion
The majority of this cohort of individuals—nearly
three quarters—presented as classic virilising CAH,
of which four experienced an additional salt-wasting
crisis.1 The proportion of individuals affected by
aldosterone deficiency causing salt-losing CAH was
less than 25%. It is generally expected for 50% to
75% of the patients with the classical form to have a
sufficiently high mineralocorticoid deficit to provoke
a salt-losing crisis.1 9 We have no explanation for why
this group of Asian individuals had such a low rate of
salt-losing crisis.
Just over a quarter of this cohort were late
presenters, which is higher than the expected 10%
to 20% late presenters described in a large case
series.2 It appears that four of these patients arrived as migrants from mainland China, explaining the
slightly large number of late presentations.
The female gender assigned at birth was
maintained by all individuals. This finding confirms
our current knowledge on 21-hydroxylase deficiency
insofar as only a small minority of CAH patients
experience gender dysphoria.2 10 It may well
be possible that some individuals in this cohort
suffered from gender issues, but a detailed interview
or questionnaire on this topic was not part of our
evaluation. A review of 250 patients identified only
5.2% suffering from gender identity problems.10
In the recent past, the concept of surgery at an early age for this group of patients has been
criticised.7 9 However, there has been an inclination
to summarise data of patients with a highly variable
background, who were operated on by a number of
different groups.11 It is now recognised that CAH
patients should be managed by a multidisciplinary
team in designated centres and the corrective
surgery may be undertaken at an early age.1 12 13
Once a decision has been reached regarding the
sex of rearing by the medical team and the parents/legal guardian, corrective surgery can be planned.
Clearly, if the clinical findings are favourable to
divert surgical intervention—as illustrated in one of
our patients—only regular review may be required
(Fig). Nevertheless, it is our impression that there
is a cultural need to decide on the sex of rearing at
an early age, as it helps to reduce the anxiety and
anguish for the family.
There is now even a tendency among
practitioners in this field to perform corrective
surgery in the first few months of life.14 It appears
that the tissue planes are easier to develop whilst
the baby is still under the influence of maternal
oestrogen effects.14
In our institution, the surgery is usually
performed in the first or second year of life. However,
four patients came to our attention late as a result
of their migration to Hong Kong. This explains the
small number of individuals in our cohort who
had their respective surgery later in life or even as
teenagers.
‘Good’ or ‘satisfactory’ results were identified
in nearly all patients in this cohort (Table 2). Other
investigators have demonstrated similar results.3 4
As identified by van der Zwan et al,15 there is a trend
for a less satisfactory outcome in patients with
high confluence. This confirms our impression
that finding high confluence poses a more difficult
challenge for the operative correction.
In our evaluation, we did not systematically
analyse behaviour traits or perform a psychological
assessment, albeit a more boyish or masculine
behaviour pattern was apparent in our cohort.
Detailed studies on this aspect of individuals with
CAH confirm this observation.16 17
All individuals sufficiently old enough to have
menses (n=15) developed a regular cycle; only two
had cycle irregularities or required medication
to support the cycle. Two patients have given
birth; elective caesarean section is recommended
after feminising genitoplasty to avoid damage to
the reconstruction; in addition, a more android
pelvic structure may result in cephalo-pelvic
disproportion.8 More detailed studies on fertility
and pregnancy conclude a reduced pregnancy and
delivery rate in women with CAH.18
Our evaluation had some limitations. Our
cohort encompassed an age-group spanning three decades. In particular, our cohort of patients did
not undergo a detailed interview process; these
offers were often declined or individuals voiced
considerable reservation to participate. Nevertheless,
this review represents the largest experience, to date,
with surgical management and the outcomes of CAH
in Asian women.
Conclusions
Nearly three quarters of our cohort presented as
classic virilising form of 21-hydroxylase deficiency.
Less than a quarter of the classic presentation
experienced an additional salt-losing crisis in this
cohort. Female gender assignment at birth was
maintained for all individuals in this group. A ‘good’
and ‘satisfactory’ outcome of surgery was reported in
nearly all patients.
Acknowledgement
We acknowledge the contributions by the Consultant
Gynaecologist, Dr Ka-wah Yiu, in the care of these
individuals.
References
1. Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal
hyperplasia due to steroid 21-hydroxylase deficiency:
an Endocrine Society clinical practice guideline. J Clin
Endocrinol Metab 2010;95:4133-60. CrossRef
2. Romao RL, Salle JL, Wherrett DK. Update on the
management of disorders of sex development. Pediatr Clin
North Am 2012;59:853-69. CrossRef
3. Farkas A, Chertin B. Feminizing genitoplasty in patients
with 46XX congenital adrenal hyperplasia. J Pediatr
Endocrinol Metab 2001;14:713-22. CrossRef
4. Rink RC, Adams MC. Feminizing genitoplasty: state of the
art. World J Urol 1998;16:212-8. CrossRef
5. Passerini-Glazel G. A new 1-stage procedure for
clitorovaginoplasty in severely masculinized female
pseudohermaphrodites. J Urol 1989;142:565-8; discussion
572.
6. Lean WL, Deshpande A, Hutson J, Grover SR. Cosmetic
and anatomic outcomes after feminizing surgery for
ambiguous genitalia. J Pediatr Surg 2005;40:1856-60. CrossRef
7. Creighton SM, Minto CL, Steele SJ. Objective cosmetic
and anatomical outcomes at adolescence of feminising
surgery for ambiguous genitalia done in childhood. Lancet
2001;358:124-5. CrossRef
8. Witchel SF. Management of CAH during pregnancy:
optimizing outcomes. Curr Opin Endocrinol Diabetes
Obes 2012;19:489-96. CrossRef
9. Ogilvie CM, Crouch NS, Rumsby G, Creighton SM, Liao
LM, Conway GS. Congenital adrenal hyperplasia in adults:
a review of medical, surgical and psychological issues. Clin
Endocrinol (Oxf) 2006;64:2-11. CrossRef
10. Dessens AB, Slijper FM, Drop SL. Gender dysphoria and
gender change in chromosomal females with congenital
adrenal hyperplasia. Arch Sex Behav 2005;34:389-97. CrossRef
11. Alizai NK, Thomas DF, Lilford RJ, Batchelor AG, Johnson N.
Feminizing genitoplasty for congenital adrenal hyperplasia:
what happens at puberty? J Urol 1999;161:1588-91. CrossRef
12. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell
WG, Speiser PW; ESPE/LWPES CAH Working Group.
Consensus statement on 21-hydroxylase deficiency from
the European Society for Paediatric Endocrinology and the
Lawson Wilkins Pediatric Endocrine Society. Horm Res
2002;58:188-95. CrossRef
13. Vidal I, Gorduza DB, Haraux E, et al. Surgical options
in disorders of sex development (dsd) with ambiguous
genitalia. Best Pract Res Clin Endocrinol Metab
2010;24:311-24. CrossRef
14. de Jong TP, Boemers TM. Neonatal management of female
intersex by clitorovaginoplasty. J Urol 1995;154:830-2. CrossRef
15. van der Zwan YG, Janssen EH, Callens N, et al. Severity
of virilization is associated with cosmetic appearance
and sexual function in women with congenital adrenal
hyperplasia: a cross-sectional study. J Sex Med 2013;10:866-75. CrossRef
16. Mathews GA, Fane BA, Conway GS, Brook CG, Hines M.
Personality and congenital adrenal hyperplasia: possible
effects of prenatal androgen exposure. Horm Behav
2009;55:285-91. CrossRef
17. Berenbaum SA, Beltz AM. Sexual differentiation of human
behavior: effects of prenatal and pubertal organizational
hormones. Front Neuroendocrinol 2011;32:183-200. CrossRef
18. Hagenfeldt K, Janson PO, Holmdahl G, et al. Fertility and
pregnancy outcome in women with congenital adrenal
hyperplasia due to 21-hydroxylase deficiency. Hum Reprod
2008;23:1607-13. CrossRef