Hong Kong Med J 2014 Oct;20(5):393–400 | Epub 25 Apr 2014
DOI: 10.12809/hkmj134126
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
The clinical utility of conventional karyotyping in the detection of cytogenetic abnormalities in soft tissue tumours: an Asian institutional experience
Justin DY Tien,1,4;
LC Lau, BSc2;
SL Tien, FAMS, FRCPA3;
MH Tan, FRCS (Edin & Glasg), FAMS1
1 Department of Orthopaedic Surgery, Singapore General Hospital, Outram Road, Singapore 169608
2 Cytogenetic Laboratory, Department of Pathology, Singapore General Hospital, Outram Road, Singapore 169608
3 Departments of Haematology and Pathology, Singapore General Hospital, Outram Road, Singapore 169608
4 School of Medicine and Biomedical Sciences, University of Sheffield,
United Kingdom
Corresponding author: Dr Justin DY Tien (juzthintien@hotmail.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
Objectives: To assess the clinical utility of
conventional karyotyping as a diagnostic tool in
soft tissue tumours amidst the increasing use of
molecular cytogenetics.
Design: Case series.
Setting: Singapore General Hospital, an Asian
institution.
Participants: A total of 35 participants (18 male and
17 female) aged 15 to 81 years were included in this
study. Conventional karyotyping of 35 consecutive
fresh soft tissue tumour specimens was performed
over 4 years and the results were analysed.
Results: Of the 35 cases of soft tissue tumours
reviewed, chromosome abnormalities were detected
in 22 (63%) cases, 11 (31%) showed a normal
karyotype, and 2 (6%) had culture failure. Of the 22
cases with abnormal karyotype, nine (41%) cases
showed recurring aberrations: Ewing’s sarcomas
(n=2), desmoplastic small round cell tumour (n=1),
synovial sarcomas (n=3), myxoid liposarcomas
(n=2), and lipoma (n=1). One lipoma case had a
t(2;12)(q23;q15) in which 2q23 breakpoint was not
reported before. Chromosomal aberration involving
12q15 breakpoint has been shown in a previous
study to be indicative of a lipoma-like liposarcoma. Another lipoma case had addition of 5q15 and 9p13
together with a balanced aberration of t(12;13)
(q13;q12) which were novel aberrations. One
synovial sarcoma case showed t(3;7)(q21;p13) which
was an uncharacteristic aberration.
Conclusion: Conventional karyotyping
demonstrated utility as a genome-wide screening
tool for soft tissue tumours and an adjunct
diagnostic tool in the event histopathology results
were doubtful. With the more widespread use
of karyotyping, novel recurring chromosomal
aberrations may be discovered.
New knowledge added by this
study
- To the authors’ knowledge, this is the first study on an Asian population documenting the clinical utility of karyotyping in the detection of cytogenetic abnormalities in soft tissue tumours. As compared with a previously published similar American cohort study which had a karyotype detection rate of 48% (n=48), this study had a higher detection rate of 63% (n=35) for chromosomal aberrations in soft tissue tumours.
- This study discovered three novel chromosomal aberration findings not previously documented before in the Mitelman Database of Chromosome Aberrations in Cancer. These comprised one lipoma, one lipoma-like liposarcoma, and one synovial sarcoma.
- This study also demonstrated the importance of karyotyping in the differential diagnosis of soft tissue tumours in cases of borderline histological results and certain cases in which the histological diagnosis did not fit the overall clinical picture.
- This study advocates the continued clinical use of conventional karyotyping as an adjunct diagnostic tool in addition to molecular cytogenetics and histology in the detection of chromosomal aberrations in soft tissue tumours. In the process, it is hoped that more novel chromosomal findings may be discovered.
Introduction
Soft tissue tumours represent a diverse group of
mesenchymal lesions which often present diagnostic
challenges to clinicians and pathologists. Histological classification of these tumours is based on their
degree of differentiation and metastatic potential:
benign, intermediate (locally aggressive), intermediate
(rarely metastasising), and malignant.1 Recent advances in molecular cytogenetics
(fluorescence in-situ hybridisation [FISH]) and
molecular assays (reverse transcription–polymerase
chain reaction [RT-PCR]) have contributed to the
ever-evolving nature of classification and diagnosis
of soft tissue sarcomas. Over the past two decades,
conventional karyotyping has demonstrated
diagnostic utility in detecting a wide range of
recurring numerical and structural chromosomal
aberrations in soft tissue tumours.
Unlike the newer molecular techniques
such as FISH, knowledge of the expected genetic
change is not required and this enables karyotyping
to function as a genome-wide screening tool.
Furthermore, karyotyping can detect any further
clonal progression in the event of a tumour
relapse. The drawbacks of karyotyping include the
dependency on sterile tumour specimens, success of
growth culture, and being time-consuming.2
Histology, immunohistochemistry, and
electron microscopy may sometimes show
borderline or non-specific features. An example
is that of malignant peripheral nerve sheath
tumours which have been historically difficult to
distinguish from other spindle cell sarcomas such
as synovial sarcomas.3 Karyotyping has shown the main difference to be the presence of the (X;18)
translocation.3 Many previous studies4 5 6 have also
demonstrated the role of conventional karyotyping
in the detection of clonal aberrations in 68% of
malignant fibrous histiocytomas, and 38 to 48%
of heterogeneous soft tissue sarcomas. Our
study aimed to highlight the use of conventional
karyotyping as a genome-wide screening tool, and
also as an adjuvant diagnostic tool in the validation
of histological diagnosis for soft tissue tumours.
Methods
Cytogenetic analysis involves a coordinated effort
between surgical pathologists and cytogenetic
laboratory technicians.6 In our study, fresh tumour
samples were collected in sterile bottles from the
surgical theatre and transported immediately to the
cytogenetics laboratory. Next, the tumour specimens
were washed 3 times with media containing Hank’s
balanced salt solution, and 2% penicillin and
streptomycin. After washing, the tissue was minced
finely with scalpels and digested in collagenase II
(GIBCO, Gaithersburg [MD], US) at a concentration
of 1400 units/mL for 1 hour. The disaggregated tissue
was then transferred into a centrifuge tube and
washed twice with 1X Hank’s balanced salt solution
and then with Roswell Park Memorial Institute complete medium (culture
medium). The cells were centrifuged and transferred
to a culture medium containing RPMI 1640, 20%
fetal bovine serum, 2% 200 mmol/L L-glutamine,
and 2% 5000 U penicillin and 5000 µg streptomycin.
Cells were cultured and harvested according to
standard cytogenetic preparations and procedures.
The cultures were set up in a 37°C incubator with 5%
CO2. The time of harvesting the cells depended on
the degree of cell proliferation in culture. At harvest,
50 µL colcemid (10 µg/mL) was added to the cultures
for 3 hours to arrest the cells at metaphase. Cultured
cells were detached by treatment with 1X trypsin
EDTA and then treated with 0.075 mol/L KCl-0.6%
trisodium citrate solution (1:2) for 20 minutes at
37°C. After fixation in two changes of methanol-acetic
acid (3:1), chromosome spreads were made by
the air-drying method. Chromosomes were stained
using the GTG banding method. A total of 20 cells
were analysed in each case and karyotype results
were designated according to International System
of Human Cytogenetic Nomenclature (ISCN 2005,
2009).7 8
Conventional karyotyping of 35 consecutive
fresh soft tissue tumour specimens was performed
in a cytogenetic laboratory at our institution over a
period of 4 years from 2005 to 2009. Medical records
and histopathology reports for each patient case
were reviewed and diagnoses were formulated based
on the World Health Organization classification
of soft tissue tumours.1 Recurrent chromosomal
abnormalities were identified using the Mitelman Database of Chromosome Aberrations in Cancer,9
and with relevant literature search. Any novel
chromosomal aberrations were also noted. This
research protocol was approved by the ethics
committee of our institution, and informed consent
from the patients was obtained by the surgeon.
Results
From January 2005 to March 2009, 35 consecutive
fresh tissue specimens were harvested from soft
tissue tumour surgical specimens. Histopathology
results revealed 20 distinct morphologies. There were
29 malignant tumours, five benign tumours, and one
of uncertain malignant potential. In our study, there
was an almost equal gender representation with 18
males and 17 females, and age ranging from 15 to
81 years. Table 1 shows an overview of the patient’s
age at diagnosis, tumour site, and tissue type for all
35 cases. The majority of our patients (37%) were
in the age-group of 41 to 60 years. The most common
tumour location was in the extremities (60%), and
adipose tissue (34%) was the most common type. As
shown in Table 2, conventional cytogenetic analysis
revealed an abnormal karyotype detection rate of
63% (22 of 35 cases). Diagnostic abnormal karyotype
was seen in nine (26%) cases—Ewing’s sarcomas
(n=2), desmoplastic small round cell tumour
(DSRCT) [n=1], synovial sarcomas (n=3), myxoid
liposarcomas (MLPSs) [n=2], and lipoma (n=1). A
normal karyotype (ie 46, XX or 46, XY) was seen
in 11 (31%) cases. There were also two (6%) cases
of culture failure. Table 3 shows the diagnosis, full karyotype results, and diagnostic utility for all 22
cases with abnormal karyotype.
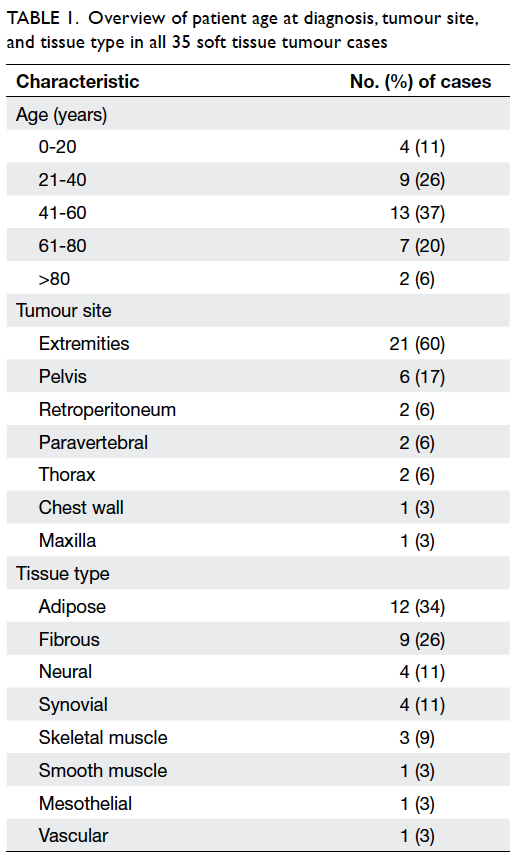
Table 1. Overview of patient age at diagnosis, tumour site, and tissue type in all 35 soft tissue tumour cases

Table 2. Detection rate of abnormal karyotype, diagnostic abnormal karyotype, normal karyotype, and culture failure in all 35 soft tissue tumour cases
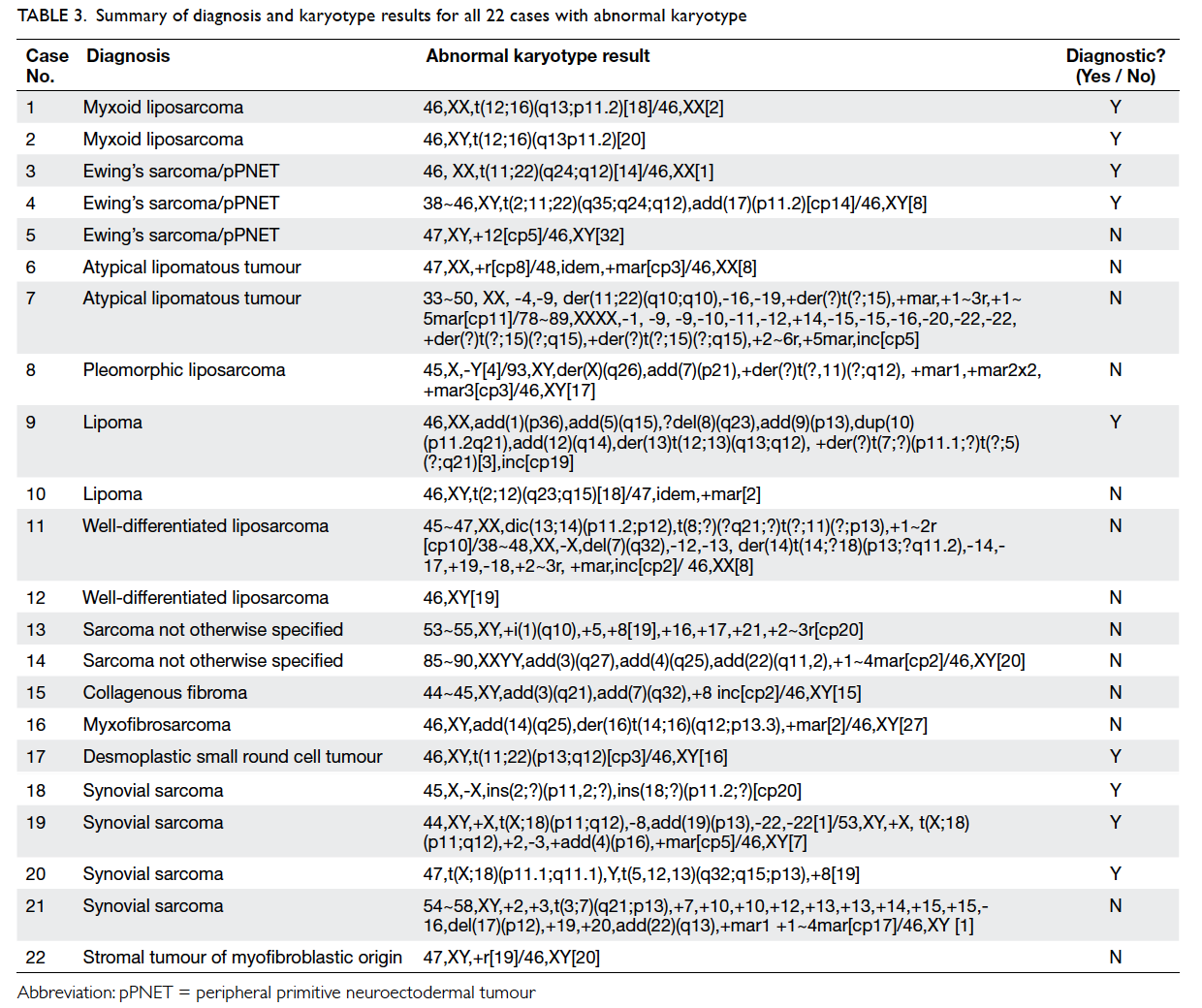
Table 3. Summary of diagnosis and karyotype results for all 22 cases with abnormal karyotype
Discussion
A wide range of structural and numerical
chromosomal abnormalities exists. These aberrations
may be characterised by chromosomal gains or losses,
balanced or unbalanced translocations, deletions or
insertions, ring or marker chromosomes, or multiple
complex karyotypes.6 Sarcomas may be categorised
into two major cytogenetic groups: (i) sarcomas
with tumour-specific chromosomal alterations and
simple karyotypes2 10 11 or (ii) sarcomas with non-specific
chromosomal alterations and complex
unbalanced karyotypes.2 For group (i), karyotypes
are considered to be tumour-specific or recurrent if
the abnormality is found in two or more cases. For
group (ii), a complex karyotype abnormality will
not be specific for the diagnosis but is supportive
of the diagnosis of malignancy. A ring chromosome
may also indicate some form of malignancy. While a
marker chromosome is diagnostically non-specific,
it is an indicator of clonal progression and further
testing by whole chromosome painting (CP) FISH
may aid the diagnosis. Chromosome painting refers to the hybridisation of fluorescently labelled
chromosome-specific probe pools for the detection
of chromosomal aberrations.12 The simultaneous
hybridisation of multiple CP probes, each tagged
with a specific fluorochrome, enables the coloured
display of all 24 human chromosomes also known
as multicolour FISH.12 The advantages of CP include
its ability to detect subtle telomeric translocations
and small chromosomal markers, barely the size of a
chromosomal band.12 Despite showing some utility as
a genetic screening tool, CP is more straightforward only when used in conjunction with conventional
cytogenetics which provide information on the
specific chromosomes involved. This is because CP
alone requires the iterative hybridisation of multiple
CP probes, which is not always practical due to time
constraints and limited specimens.12
Of the 22 cases with abnormal karyotype
results, nine (41%) cases showed tumour-specific
chromosome abnormalities. These nine cases had
abnormal karyotypes which were consistent with the
Mitelman Database of Chromosome Aberrations in
Cancer9 and previously published literature.2 11 A
normal karyotype was seen in 11 (31%) cases where
three tumour tissues were of fibrous origin. One
study in our literature search demonstrated a normal
karyotype in 42% of cases; the majority of these were
soft tissue tumours with a fibrous component or grossly dense matrix.6 The study rationalised that
tumour cells embedded in a dense matrix were more
difficult to culture.6 The two culture failure cases
could have been due to specimen contamination
or insufficient sample. A study conducted in a
single institution in the United States (n=48) had
documented an abnormal karyotype detection rate
of 48% and a 10% culture failure rate in patients
with soft tissue tumours.6 In contrast, our Asian
cohort study had a higher detection rate of 63%
(n=35) and a lower culture failure rate of 6%. The
small sample size of this study was limited by the
disease prevalence (rarity of sarcomas) as well as
the logistics of obtaining fresh specimens from the
surgical operating room. We intend to conduct
future studies with a bigger sample size and explore
other cytogenetic aberrations in soft tissue sarcomas using FISH in conjunction with conventional
karyotyping.
Ewing’s sarcoma/peripheral primitive neuroectodermal tumour
Of the two cases of Ewing’s sarcoma in this study,
one was a 42-year-old female (case 3; Table 3) and
one a 26-year-old male (case 4; Table 3). This is
an unusual clinical age-group for this sarcoma
and the histological diagnosis was confirmed
by karyotyping. In the male patient, the variant
t(2;11;22)(q35;q24;q12) was demonstrated. For case
5 (Table 3), trisomy 12, a non-random secondary
aberration, was demonstrated. One study found that
the majority of chromosomal aberrations in Ewing’s
sarcoma appear to be trisomy 8 and trisomy 12,
occurring in 44% and 16% of Ewing’s sarcoma cases,
respectively.13 14 15
Synovial sarcoma
Our study showed two diagnostic cases of synovial
sarcoma (cases 19 and 20) with the hallmark
translocation t(X;18) seen16 together with complex
cytogenetic aberrations (Table 3). Another two cases of synovial sarcoma (cases 18 and 21) showed structure rearrangement on 2p/18p and translocation t(3;7)(q21;p13), respectively. These abnormalities were uncharacteristic. Histological biopsy
of the left distal tibia showed a soft tissue tumour
measuring 4 x 3 x 1 cm, composed of large sheets of
malignant cells displaying high nuclear cytoplasmic
ratio, round or irregular nuclei, nucleoli, scanty
cytoplasm, and frequent mitoses. Tumour cells
were positive for CD99 (MIC2 gene product),
cytokeratin AE1+3 (especially epithelial-like areas),
and vimentin. Further immunohistochemical
staining with epithelial membrane antigen showed
focal positivity. The soft tissue tumour had also
invaded the distal tibia on the anteromedial and
posteromedial aspects of the left leg with metastasis
to the left groin lymph node. Case 21 was reviewed by
various histopathologists and the general consensus
was that of a high-grade undifferentiated synovial
sarcoma. The representative karyogram for case 21
is shown in Figure 1.
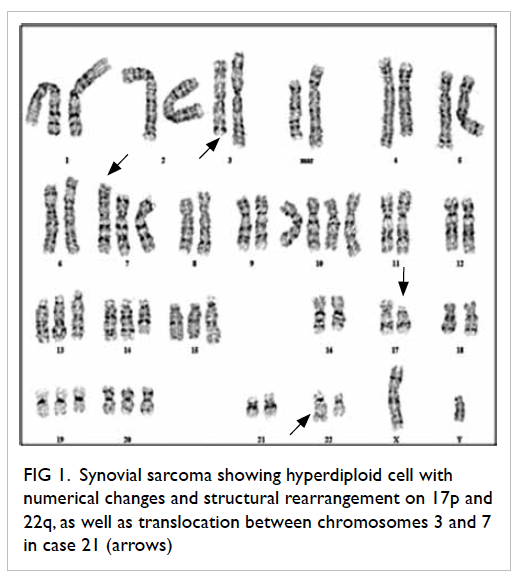
Figure 1. Synovial sarcoma showing hyperdiploid cell with numerical changes and structural rearrangement on 17p and 22q, as well as translocation between chromosomes 3 and 7 in case 21 (arrows)
In the study by Saboorian et al,17 there was one
case of ambiguous histological results; the stained
tissue smears showed densely cellular and tightly
cohesive malignant spindle cells without discernible
epithelial differentiation. A few differential
diagnoses were formulated which included synovial
sarcoma and karyotyping confirmed the diagnosis
of synovial sarcoma by revealing the presence of
t(X;18)(p11.2;q11.2).17 Another study by Akerman
et al,18 which involved the cytogenetic evaluation
of 15 surgical specimens, confirmed the (X;18)
translocation as both a specific and sensitive marker
for synovial sarcoma. Our study and the above
studies serve to highlight the essential supportive role of conventional karyotyping in the confirmation
of the diagnosis of synovial sarcoma.
Liposarcoma
Liposarcoma is the most common soft tissue sarcoma,
accounting for 20% of mesenchymal neoplasms.19 It
can be categorised into three subtypes: myxoid and
round cell, well-differentiated, and pleomorphic.19
All three subtypes that were included in our study
are discussed below.
Myxoid liposarcoma
Myxoid liposarcoma is the second most common
liposarcoma subtype in which two thirds of the
cases arise from the thigh musculature.19 The
characteristic translocation t(12;16)(q13;p11) has
been well documented in more than 90% of MLPS
cases.19 20 21 22 This translocation leads to formation of a
TLS-CHOP fusion gene (located at 12q13 and 16p11
respectively) which is highly sensitive and specific
for MLPS.19 A possible trisomy 8 as an additional
secondary change has also been reported.22 Our
study demonstrated two cases of MLPS showing
the t(12;16)(q13;p11) translocation. As shown in
Table 3, this translocation was diagnostic of MLPS
in cases 1 and 2. A study by the CHAMP group in
which cytogenetic analysis was carried out in 28
MLPS specimens reported the t(12;16)(q13;p11)
translocation in 26 cases; this further confirmed
its consistency as a genetic marker for MLPS.20
Conventional karyotyping for t(12;16)(q13;p11) in
MLPS was also shown to be useful as an adjunct
diagnostic tool in poorly differentiated myxoid
neoplasms in another study.21
Pleomorphic liposarcoma
Pleomorphic liposarcoma (PLPS) is the rarest
(5% of liposarcoma) and most aggressive (highly
metastatic) form of liposarcoma.19 It commonly
affects the extremities in the elderly (>50 years)
with an equal distribution in both genders.19 The
complex structural abnormalities (unidentified
marker chromosomes) and high chromosome
counts (polyploidy) make it difficult to detect PLPS-specific
aberrations.19 Our study demonstrated the
case of a 77-year-old female (case 8; Table 3) with
PLPS which showed complex, structural aberrations
on karyotyping which, though not diagnostic, was
indicative of a malignant clonal process.
Lipoma
Lipomas are the most common soft tissue tumours
and are benign.23 One study by Sandberg and Bridge24
had documented rearrangements affecting the
12q13~q15 region as the most common aberration
(65% of 188 lipomas). Clonal chromosomal
aberrations were also reported in 60% of lipomas,
and of these, 70% had normal cytogenetic cells.24
The most frequent t(3;12)(q27~q28;q13~q15)
translocation was seen in 25% of lipoma cases.24
Case 10 (Table 3) belonging to a 53-year-old
male demonstrated the balanced t(2;12)
(q23;q15) translocation which was also novel in
that the breakpoint 2q23 has not been previously
reported. In this patient, histology showed a large
lipoma measuring 14 x 9 x 8 cm. In addition,
magnetic resonance imaging suggested a malignant
liposarcoma. Szymanska et al25 found that the
overrepresentation of 1q and 12q sequences was a
recurrent finding in lipoma-like liposarcomas but not
in lipomas. This is consistent with the chromosomal
aberration involving 12q15 breakpoint in case 10.
The representative karyogram for case 10 is shown
in Figure 2.
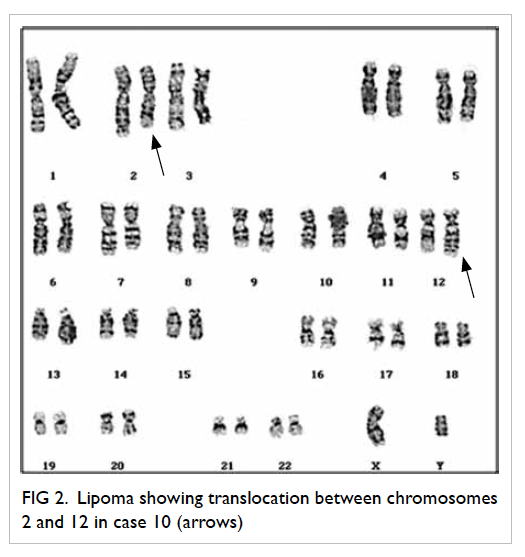
Figure 2. Lipoma showing translocation between chromosomes 2 and 12 in case 10 (arrows)
Atypical lipomatous tumour/well-differentiated
liposarcoma
Atypical lipomatous tumour (ALT) is synonymous
with well-differentiated liposarcoma (WDLPS) as
both exhibit similar cytogenetic findings regardless
of location and pathology.19 Being the most common
of all liposarcomas (40%-45%), ALT/WDLPS is
an intermediate (locally aggressive) soft tissue
sarcoma with mature adipocyte differentiation.1 19 26
Most ALTs are characterised cytogenetically by the
presence of supernumerary ring chromosomes or
long marker chromosomes involving chromosome
region 12q13-15.26 27
Our study demonstrated abnormal karyotypes
in two cases of ALT and WDLPS each. Of the two
ALTs, case 6 (Table 3) had a ring chromosome as a
sole abnormality and case 7 had supernumerary ring chromosomes present in addition to the multiple
complex numerical structural aberrations. Case
11 (WDLPS) showed both complex numerical
and structural chromosomal rearrangements in
which two dicentric chromosomes were present
together with ring chromosomes and giant marker
chromosomes. Case 12 (WDLPS) belonged to a
65-year-old male; a normal karyotype was seen in 19
cells, one nonclonal abnormal cell was hypodiploid
which showed trisomy 12, deletion on 12p, structural
rearrangement on 20q as well as a ring chromosome.
It is uncertain if this nonclonal abnormal cell is of
any clinical significance. Histology had showed a
WDLPS measuring 19 x 12 x 4 cm infiltrating the
skeletal muscle of the left thigh.
It was reported that virtually all ALT/WDLPS
had abnormal cytogenetic results.26 The CHAMP
group conducted a study of 59 ALT/WDLPS
and evaluated their relationship and differential
diagnoses with other adipose tissue tumours.28
Clonal chromosomal abnormalities were found in 55
(93%) cases and supernumerary ring or giant marker
chromosomes (RGCs) were seen in 37 (63%) cases28;
RGCs were also shown to have tumour progression
potential. Statistical analysis demonstrated a highly
significant correlation between ALTs and RGCs
(P<0.0001).28 The study reaffirmed the essential role
of karyotype analysis in differentiating ALTs from
benign lipomas, spindle/PLPS, hibernomas, and
MLPS.
Desmoplastic small round cell tumour
Desmoplastic small round cell tumour is a rare
and aggressive neoplasm that commonly affects adolescents and young adults.29 30 Our study
demonstrated the case of a 27-year-old male with
DSRCT showing the classic t(11;22)(p13;q12)
translocation (case 17; Table 3). In this case,
histopathology reports showed no evidence of
malignant infiltrates in the tumour specimen but
conventional karyotyping confirmed the diagnosis
to be DSRCT.
Conclusion
Karyotype analysis detected a majority (63%) of cases
with abnormal chromosomes in our Asian cohort
study with nine (41%) cases showing 22 abnormal
karyotypes. Our study, hence, demonstrated that
conventional karyotyping played an essential
supportive role in validating histological diagnosis,
especially in cases with borderline or complex
morphology. Newer molecular techniques such as
FISH and RT-PCR techniques may be sensitive but
require prior knowledge of the expected genetic
change. In view of this, conventional karyotyping is
useful as a genome-wide screening tool in detecting
single or multiple chromosomal aberrations in each
patient. The use of conventional karyotyping is highly
encouraged in the pursuit of discovering more novel
recurring chromosomal aberrations.
Acknowledgements
This study was internally supported by the grant
from the Department of Clinical Research and
Cytogenetics Laboratory, Department of Pathology,
Singapore General Hospital. The authors would like
to thank Dr Alvin Lim for his support and Mr Lim
Ping for his technical assistance in ensuring the
success of this research project.
References
1. Fletcher CD, Unni KK, Mertens F, editors. World Health Organization classification of tumors: pathology and genetics of tumors of soft tissue and bone. Lyon, France: IARC Press; 2002.
2. Antonescu CR. The role of genetic testing in soft-tissue sarcoma. Histopathology 2006;48:13-21. CrossRef
3. Fletcher JA, Kozakewich HP, Hoffer FA, et al. Diagnostic relevance of clonal cytogenetic aberrations in malignant soft-tissue tumours. N Engl J Med 1991;324:436-43. CrossRef
4. Mandahl N, Heim S, Wilen H, et al. Characteristic karyotypic anomalies identify subtypes of malignant fibrous histiocytoma. Genes Chromosomes Cancer 1989;1:9-14. CrossRef
5. Becher R, Wake N, Gibas Z, Ochi H, Sandberg AA. Chromosome changes in soft-tissue sarcomas. J Natl Cancer Inst 1984;72:823-31.
6. Matthews A, Tang M, Cooper K. Cytogenetic aberrations in soft tissue tumours harvested from fresh tissue submitted for surgical pathology: a single institution experience. Int J Surg Pathol 2010;18:260-7. CrossRef
7. Schaffer LG, Tommerup N, editors. ISCN 2005: an international system for human cytogenetic nomenclature. Basel, Switzerland: S Karger; 2005.
8. Schaffer LG, Slovak ML, Campbell LJ, editors. ISCN 2009: an international system for human cytogenetic nomenclature. Basel, Switzerland: S Karger; 2009.
9. Cancer Genome Anatomy Project. Mitelman database of chromosome aberrations in cancer. Available from: http://cgap.nci.nih.gov/Chromosomes/RecurrentAberrations. Updated August 14, 2009. Accessed 25 Aug 2009.
10. Sreekantaiah C, Ladanyi M, Rodriguez E, Sreekantaiah C. Chromosomal aberrations in soft tissue tumors. Relevance to diagnosis, classification and molecular mechanisms. Am J Pathol 1994;144:1121-34.
11. Dei Tos AP, Dal Cin P. The role of cytogenetics in the classification of soft tissue tumours. Virchows Arch 1997;431:83-94. CrossRef
12. Reid T, Schrock E, Ning Y, Wienberg J. Chromosome painting: a useful art. Hum Mol Genet 1998;7:1619-26. CrossRef
13. Roberts P, Burchill SA, Brownhill S, et al. Ploidy and karyotype complexity are powerful prognostic indicators in the Ewing’s sarcoma family of tumors: a study by the United Kingdom cancer cytogenetics and the children’s cancer and leukaemia group. Genes Chromosomes Cancer 2008;47:207-20. CrossRef
14. Mugneret F, Lizard S, Aurias A, Turc-Carel C. Chromosomes in Ewing’s sarcoma. II. Nonrandom additional changes, trisomy 8 and der(16)t(1;16). Cancer Genet Cytogenet 1988;32:239-45. CrossRef
15. Maurici D, Perez-Atayde A, Grier HE, Baldini N, Serra M, Fletcher JA. Frequency and implications of chromosome 8 and 12 gains in Ewing’s sarcoma. Cancer Genet Cytogenet 1998;100:106-10. CrossRef
16. Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumours. Synovial sarcoma. Cancer Genet Cytogenet 2002;133:1-23. CrossRef
17. Saboorian MH, Ashfaq R, Vandersteenhoven JJ, Schneider NR. Cytogenetics as an adjunct in establishing a definitive diagnosis of synovial sarcoma by fine-needle aspiration. Cancer 1997;81:187-92. CrossRef
18. Akerman M, Willén H, Carlén B, Mandahl N, Mertens F. Fine needle aspiration (FNA) of synovial sarcoma: a comparative histological-cytological study of 15 cases, including immunohistochemical, electron microscopic and cytogenetic examination and DNA-ploidy analysis. Cytopathology 1996;7:187-200. CrossRef
19. Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft-tissue tumours: liposarcoma. Cancer Genet Cytogenet 2004;155:1-24. CrossRef
20. Tallini G, Akerman M, Dal Cin P, et al. Combined morphologic and karyotypic study of 28 myxoid liposarcomas. Implications for a revised morphologic typing (a report from the CHAMP Group). Am J Surg Pathol 1996;20:1047-55. CrossRef
21. Ohjimi Y, Iwasaki H, Ishiguro M, et al. Myxoid liposarcoma with t(12;16)(q13; p11). Possible usefulness of chromosome analysis in a poorly differentiated sarcoma. Pathol Res Pract 1992;188:736-41. CrossRef
22. Sreekantaiah C, Karakousis CP, Leong SP, Sandberg AA. Trisomy 8 as a non-random secondary change in myxoid liposarcoma. Cancer Genet Cytogent 1991;51:195-205. CrossRef
23. Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft-tissue tumors: lipoma. Cancer Genet Cytogenet 2004;150:93-115. CrossRef
24. Sandberg AA, Bridge JA. The cytogenetics of bone and soft tissue tumours. Austin: RG Landes; 1994.
25. Szymanska J, Virolainen M, Tarkkanen M, et al. Overrepresentation of 1q21-23 and 12q13-21 in lipoma-like liposarcomas but not in benign lipomas: a comparative genomic hybridization study. Cancer Genet Cytogenet 1997;99:14-8. CrossRef
26. Dei Tos AP, Doglioni C, Piccinin S, et al. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumours. J Pathol 2000;190:531-6. CrossRef
27. Rubin BP, Fletcher CD. The cytogenetics of lipomatous tumours. Histopathology 1997;30:507-11. CrossRef
28. Rosai J, Akerman M, Dal Cin P, et al. Combined morphologic and karyotypic study of 59 atypical lipomatous tumours. Evaluation of their relationship and differential diagnosis with other adipose tissue tumours (a report of the CHAMP Study Group). Am J Surg Pathol 1996;20:1182-9. CrossRef
29. Gerald WL, Haber DA. The EWS-WT1 gene fusion in desmoplastic small round cell tumor. Semin Cancer Biol 2005;15:197-205. CrossRef
30. Lae ME, Roche PC, Jin L, Lloyd RV, Nascimento AG. Desmoplastic small round cell tumour: a clinicopathologic, immunohistochemical and molecular study of 32 tumours. Am J Surg Pathol 2002;26:823-35. CrossRef