DOI: 10.12809/hkmj134158
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
An uncommon cause of Cushing’s syndrome in a 70-year-old man
Kitty KT Cheung, MRCP, FHKAM (Medicine)1; WY So, FRCP, FHKAM (Medicine)1; Alice PS Kong, FRCP, FHKAM (Medicine)1; Ronald CW Ma, FRCP, FHKAM (Medicine)1; KF Lee, FRCSEd (Gen), FHKAM (Surgery)2; Francis CC Chow, FRCP, FHKAM (Medicine)1
1 Department of Medicine and Therapeutics, The Chinese University of Hong Kong, Prince of Wales Hospital, Shatin, Hong Kong
2 Department of Surgery, The Chinese University of Hong Kong, Prince of Wales Hospital, Shatin, Hong Kong
Corresponding author: Dr Kitty KT Cheung (kittyktcheung@cuhk.edu.hk)
 Full
paper in PDF
Full
paper in PDF
Abstract
Cushing’s syndrome due to exogenous steroids is
common, as about 1% of the general populations
use exogenous steroids for various indications.
Although endogenous Cushing’s syndrome due
to ectopic adrenocorticotropic hormone from a
pancreatic neuroendocrine tumour is rare, a correct
and early diagnosis is important. The diagnosis
and management require high clinical acumen
and collaboration between different specialists.
We report a case of ectopic adrenocorticotropic
hormone Cushing’s syndrome due to pancreatic
neuroendocrine tumour with liver metastasis. Early
recognition by endocrinologists with timely surgical
resection followed by referral to oncologists led to a
favourable outcome for the patient up to 12 months after initial presentation.
Case report
In March 2013, a 70-year-old Chinese man presented
with polyuria and polydipsia was diagnosed to
have new-onset type 2 diabetes mellitus. He had
suboptimal glycaemic control, and received multiple
oral hypoglycaemic agents (OHAs). At the same
time, he was noted to have bilateral lower limb
pitting oedema and difficult to heal wounds over
feet, as well as persistent hypokalaemia for which he
was prescribed regular treatment with a potassium-sparing diuretic and oral potassium supplements.
Symptoms and signs of Cushing’s syndrome (CS)
including easy bruising, proximal muscle weakness,
and central obesity were subsequently detected
(Fig 1). He denied any history of taking herbal
medicine or exogenous steroids. The overnight
1-mg dexamethasone screening test for CS yielded
a non-suppressible plasma cortisol level of 1308
(reference level [RL], <50) nmol/L. Paired 9am cortisol
and adrenocorticotropic hormone (ACTH) levels were 1220 nmol/L and 78 pmol/L (RL, <10.2
pmol/L), respectively. Two sets of values for 24-hour
urinary free cortisol excretion were strikingly
high at 2263 and 3601 nmol/day (reference range, 35-151 nmol/day). He also failed the confirmatory
low-dose dexamethasone suppression test with a
cortisol level of 997 nmol/L (RL, <50 nmol/L) after
2 days of dexamethasone loading. The peripheral
corticotropin-releasing hormone (CRH) stimulation
test later established the diagnosis of ectopic ACTH
CS, since both the ACTH and cortisol responses
were flat after CRH injection. Ketoconazole was
commenced at that juncture, which was 2 months
after the patient’s initial presentation.
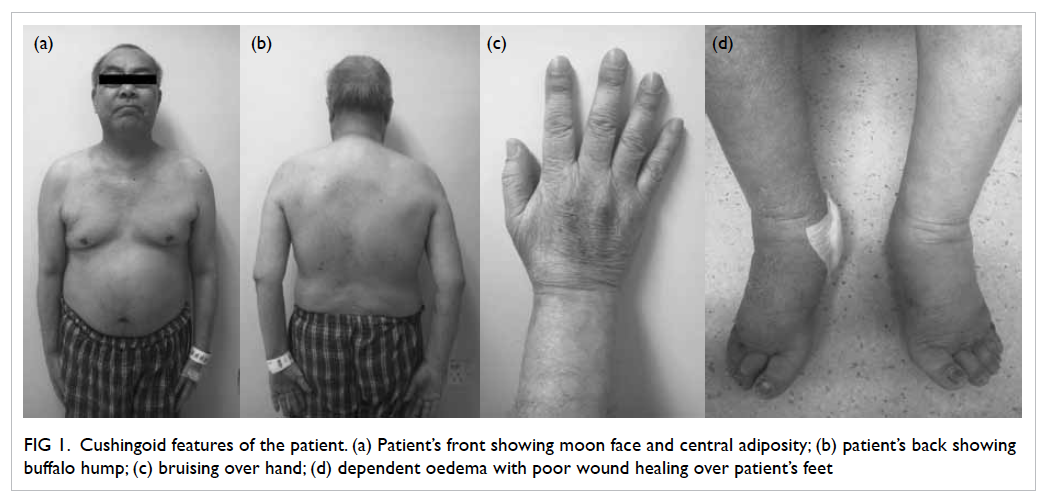
Figure 1. Cushingoid features of the patient. (a) Patient’s front showing moon face and central adiposity; (b) patient’s back showing buffalo hump; (c) bruising over hand; (d) dependent oedema with poor wound healing over patient’s feet
Contrast computed tomography (CT) of the
thorax and abdomen followed immediately, and
revealed a well-defined ovoid cystic area (6.3 x 4.8
x 4.9 cm) with an intralesional eccentric isodense
mildly enhanced mural nodule in the body of pancreas
that was consistent with pancreatic tumour, with
enlarged lymph nodes posterior to the body of the
organ (Figs 2a and 2b). Mild generalised osteoporosis
was also noted. Positron emission tomography (PET)
of the whole body 2 weeks later showed a mildly hypermetabolic heterogeneous lesion in the body of
the pancreas, compatible with the known pancreatic
tumour. Also, there were mildly hypermetabolic
lymph nodes in the peripancreatic region, possibly
due to early nodal involvement. The serum CA19.9 level (a tumour marker of pancreatic cancer) was
elevated (89 kIU/L; RL, <18 kIU/L).
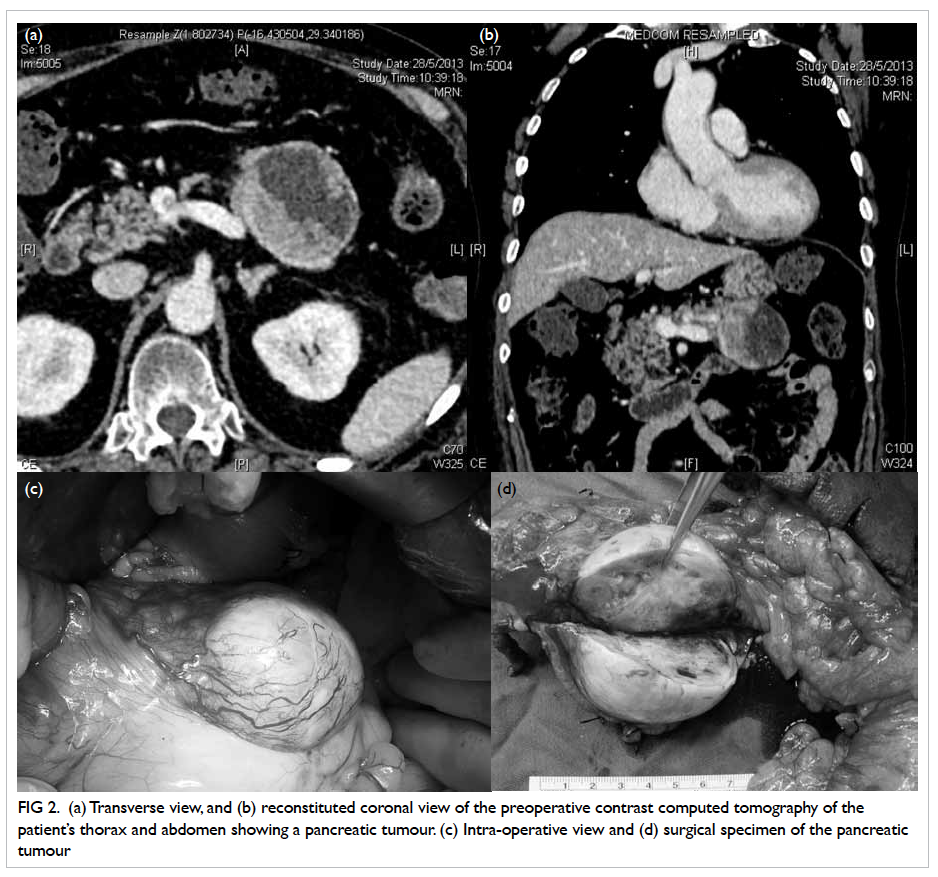
Figure 2. (a) Transverse view, and (b) reconstituted coronal view of the preoperative contrast computed tomography of the patient’s thorax and abdomen showing a pancreatic tumour. (c) Intra-operative view and (d) surgical specimen of the pancreatic tumour
The patient was referred to surgeons 3
months after initial presentation, and offered distal
pancreatectomy with splenectomy (Figs 2c and 2d).
Intra-operatively, a solitary 2-mm nodule over the
undersurface of segment III of liver, not identified
in the preoperative CT, was found and histologically
confirmed to be metastatic neuroendocrine tumour
(NET). Intra-operative ultrasound did not reveal
any other liver lesions. There were no palpable
lesions over whole length of small bowel or colon
in the peritoneum or the omentum. Histology
of the resected pancreatic mass confirmed the
presence of malignant pancreatic NET (P-NET)
with extrapancreatic extension and lymphovascular
permeation. The tumour cells were diffusely positive
for CK19, synaptophysin, and chromogranin.
Staining for ACTH, gastrin, and pancreatic
polypeptidase were focally positive, but staining for
insulin, serotonin, somatostatin, and glucagon were
all negative. The proliferative pool as assessed by
Ki-67 was estimated to be approximately 15%.
Postoperatively, ketoconazole was stopped,
and the patient started taking replacement doses
of hydrocortisone. He was then referred to an
oncologist for further management in view of the
metastatic nature of his disease (stage IV P-NET due
to confirmed liver metastasis). One month after the
operation, the patient experienced marked alleviation
of his symptoms. He had no more oedema and the
OHA requirements were significantly reduced.
Discussion
Cushing’s syndrome due to exogenous steroids is
common, as about 1% of the general populations use
exogenous steroids for various indications.1 Ectopic
ACTH secretion accounts for approximately 10% to
20% of all cases of CS.2 The leading cause is small-cell
lung carcinoma, accounting for about 50% of
the cases. Other less common tumours reported
are pancreatic, bronchial, thymic, and thyroid
medullary carcinoma. Certainly, P-NETs are rare
and have an incidence of approximately 1/100 000
persons per year, and both genders appear equally
prone.3 Among the P-NETs, insulinoma, gastrinoma,
glucagonoma, somatostatinoma, and VIPoma have
all been reported. Other non-functioning islet
neoplasms and other hormone-secreting (eg ACTH)
tumours have also been published in case reports.4
Other than insulinoma, these P-NETs are generally
malignant. Those that are ACTH-producing
(account for approximately 1.2% of them) are particularly aggressive.4 Metastases, usually to the
liver, are often observed in early phase, even before
the presentation of CS.5 The 2- and 5-year survival
rates of patients with P-NETs are about 40% and
16%, respectively.6
Symptoms and signs from excess cortisol,
followed by biochemical evaluation and subsequent
imaging, as in our patient, are important in the timely
diagnosis of functioning P-NETs. In our patient,
both the screening and other confirmatory tests for
CS established the diagnosis. Non-suppressible/high
ACTH in the presence of high serum concentrations
and urinary secretion of cortisol, coupled with flat
ACTH and cortisol responses after provocative
peripheral CRH stimulation test, strongly suggested
the CS was due to an ectopic ACTH-secreting source
rather than the pituitary.
Other than the peripheral CRH stimulation test
which offers 86% sensitivity and 90% specificity for
pituitary CS,7 high-dose dexamethasone suppression
test (HDDST) and bilateral inferior petrosal sinus
(IPS) sampling for ACTH are two other options for
differentiating pituitary CS and ectopic ACTH CS.
A positive HDDST, characterised by suppression
of serum cortisol by ≥50% from baseline by 8 mg
of dexamethasone taken at 11 pm the night before,
offers 77% sensitivity and 60% specificity for CS.
The rationale for the use of HDDST is based on the
principle that pituitary tumours are only partially
autonomous, retaining feedback mechanism at a
higher set point than normal. Therefore, when enough
dexamethasone is administered, ACTH and cortisol
secretion can be suppressed. While for ectopic
ACTH tumours, which are usually autonomous,
production of hormones cannot be suppressed with
dexamethasone. However, some benign ectopic
tumours may be suppressible, while pituitary
macroadenomas are often non-suppressible.7 Whilst
IPS sampling is invasive, it is the most direct way
to examine whether the pituitary is the source of
excess ACTH. An IPS/periphery ACTH ratio of >2.0
correctly identifies CS with 95% sensitivity and 100%
specificity. The sensitivity is further improved to
100% when CRH is administered using the cut-off of
post-CRH IPS/periphery ratio of >3.0.8
In our case, immediate search for the ACTH-secreting
source using CT and PET identified
the pancreatic tumour promptly. Other imaging
modalities commonly used in localising NETs
include magnetic resonance imaging, endoscopic
ultrasound, and somatostatin receptor scintigraphy.
The source of ACTH in 30% to 50% of patients
with ACTH-dependent CS is not localised by the
conventional imaging modalities listed above.9
Newer imaging techniques such as fluorine-labelled
dihydroxyphenylalanine (18F-DOPA) PET/CT are
now being used to localise occult sources, although
the usefulness of some of them remains controversial. In a series of 17 patients, no advantage was seen
with tumour localisation using (18F-DOPA) PET/CT
when compared with conventional imaging, while
another study reported 100% localisation of ectopic
ACTH-secreting NETs using (18F-DOPA) PET/CT
in three patients.9 10
Treatments for P-NETs include surgery,
chemotherapy, radiotherapy, and interventional
radiology techniques such as hepatic artery chemoembolisation.
Surgery is the first-line option for
resectable tumours and is also used for debulking
metastatic tumours. Total hepatectomy with living
donor transplantation has also been attempted
for treating metastatic tumours.11 Somatostatin
and its analogues have both antisecretory and
antiproliferative effects.12 Although P-NETs are
relatively radioresistant, recently developed peptide
receptor radiotherapy employing radionuclide-targeted
somatostatin receptor agonists for internal
cytotoxic radiotherapy in somatostatin receptor-expressing
NETs seem promising.12 Systemic
therapies for unresectable tumours include sunitinib
malate, a potent tyrosine kinase inhibitor with
antiangiogenic effects, and everolimus, an inhibitor
of mammalian target of rapamycin.12 13 After surgical
resection of malignant P-NETs, Ki-67 >5% of tumour
cells is a predictor of recurrence.5 Since our patient
had a Ki-67 of approximately 15%, oncological
treatment will be needed, hence, the referral.
In conclusion, our patient with an ectopic
ACTH-secreting P-NET presented with diabetes and
hypertension, both of which are common chronic
diseases worldwide. Due to the aggressive nature of
this type of tumour and its histological findings, this
patient will likely require further adjuvant treatments
in the future. Ectopic ACTH CS can occur due to
a wide spectrum of causes, and a combination of
relevant biochemical tests and imaging are needed
to establish the correct diagnosis. Timely referral to
surgeons and/or oncologists is necessary. Symptoms
of hormone excess are often the first hint suggesting
the diagnosis of functioning P-NETs. Almost all
P-NETs, except insulinoma, carry a high malignant
potential. Expeditious and meticulous management
involving collaboration between endocrinologists,
surgeons, pathologists, and oncologists can be
expected to provide the best outcomes for patients suffering from this rare disease.
References
1. Prague JK, May S, Whitelaw BC. Cushing’s syndrome. BMJ
2013;346:f945. CrossRef
2. Wajchenberg BL, Mendonca BB, Liberman B, et al. Ectopic
adrenocorticotropic hormone syndrome. Endocr Rev
1994;15:752-87. CrossRef
3. Eriksson B, Oberg K. Neuroendocrine tumours of the
pancreas. Br J Surg 2000;87:129-31. CrossRef
4. Ito T, Tanaka M, Sasano H, et al. Preliminary results
of a Japanese nationwide survey of neuroendocrine
gastrointestinal tumors. J Gastroenterol 2007;42:497-500. CrossRef
5. Doppman JL, Nieman LK, Cutler GB Jr, et al.
Adrenocorticotropic hormone–secreting islet cell tumors:
are they always malignant? Radiology 1994;190:59-64. CrossRef
6. Clark ES, Carney JA. Pancreatic islet cell tumor associated
with Cushing’s syndrome. Am J Surg Pathol 1984;8:917-24. CrossRef
7. Reimondo G, Paccotti P, Minetto M, et al. The
corticotrophin-releasing hormone test is the most reliable
noninvasive method to differentiate pituitary from ectopic
ACTH secretion in Cushing’s syndrome. Clin Endocrinol
(Oxf) 2003;58:718-24. CrossRef
8. Invitti C, Pecori Giraldi F, de Martin M, Cavagnini F.
Diagnosis and management of Cushing’s syndrome: results
of an Italian multicentre study. Study Group of the Italian
Society of Endocrinology on the Pathophysiology of the
Hypothalamic-Pituitary-Adrenal Axis. J Clin Endocrinol
Metab 1999;84:440-8. CrossRef
9. Pacak K, Ilias I, Chen CC, Carrasquillo JA, Whatley
M, Nieman LK. The role of [(18)F]fluorodeoxyglucose
positron emission tomography and [(111)In]-diethylenetriaminepentaacetate-D-Phe-pentetreotide
scintigraphy in the localization of ectopic
adrenocorticotropin-secreting tumors causing Cushing’s
syndrome. J Clin Endocrinol Metab 2004;89:2214-21. CrossRef
10. Kumar J, Spring M, Carroll PV, Barrington SF, Powrie JK.
18Flurodeoxyglucose positron emission tomography in the
localization of ectopic ACTH-secreting neuroendocrine
tumours. Clin Endocrinol (Oxf) 2006;64:371-4.
11. Blonski WC, Reddy KR, Shaked A, Siegelman E, Metz
DC. Liver transplantation for metastatic neuroendocrine
tumor: a case report and review of the literature. World J
Gastroenterol 2005;11:7676-83.
12. Wiedenmann B, Pavel M, Kos-Kudla B. From targets to
treatments: a review of molecular targets in pancreatic
neuroendocrine tumors. Neuroendocrinology
2011;94:177-90. CrossRef
13. Hörsch D, Grabowski P, Schneider CP, et al. Current
treatment options for neuroendocrine tumors. Drugs
Today (Barc) 2011;47:773-86.