Hong Kong Med J 2025 Feb;31(1):75–7 | Epub 17 Feb 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
COMMENTARY
Novel LMX1B variants in nail-patella syndrome
LT Leung, MB, BS#; Stephanie KL Ho, MB, ChB#; WC Yiu, MSc; Shirley SW Cheng, MB, ChB; Ivan FM Lo, MB, ChB, HM Luk, MD
Department of Clinical Genetics, Hong Kong Children’s Hospital, Hong Kong SAR, China
# Equal contribution
Corresponding author: Dr HM Luk (lukhm@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Nail-patella syndrome (NPS, OMIM #161200) is a
rare autosomal dominant multisystem disorder with
an estimated prevalence of 1 in 50 000 population.1
Despite its complete penetrance, underdiagnosis
is suspected due to highly variable expressivity;
presentations range from clinically significant
skeletal anomalies accompanied by end-stage renal
disease to isolated nail dystrophy. The achievement
of a molecular diagnosis has broad implications for
surveillance and management because ophthalmic
and gastrointestinal involvement have been reported
in association with NPS.1
In this commentary, we summarised the
clinical and molecular characteristics of seven cases
with molecularly confirmed NPS from five unrelated
families at our hospital. The affected individuals
ranged from 11 years to 73 years. All individuals in
our cohort were male, except for Individual 2 (male-to-female ratio=6:1). A positive family history, with
at least one clinically affected relative, was identified
for each individual except Individual 5, who was
subsequently confirmed to harbour a de novo
LMX1B (LIM homeobox transcription factor 1 beta)
variant.
Nail involvement was present in almost all
affected individuals (6/7, 85.7%). Nail dystrophy
with bilateral involvement, the most common
manifestation, was associated with varying degrees
of ridging and splitting. Triangular lunulae were
identified in two affected individuals (28.6%), while
knee involvement was observed in four affected
individuals (57.1%). Three individuals (42.9%)
displayed bilaterally small patellae, two of whom
experienced complications with subluxation, whereas
one exhibited bilaterally absent patellae. Elbow
involvement was evident in only two individuals: one
presented with bilateral elbow flexion contracture
deformity and the other showed limited bilateral
elbow movement. Bilateral iliac horns were detected
in three affected individuals. Renal involvement
was identified in only one individual (14.3%), who
exhibited microscopic haematuria; the remaining
individuals showed no renal manifestations. Three
individuals presented with ocular manifestations.
Notably, Individual 2 displayed ptosis and corneal
opacity at birth; she was subsequently diagnosed with
bilateral congenital ectropion uvea with glaucoma,
which was complicated by high myopia and lattice
degeneration of the retina in the right eye and
rhegmatogenous retinal detachment in the left eye.
Her father (Individual 3) had experienced blindness
in his right eye since birth, as well as bilateral
glaucoma. Individual 7 had underlying bilateral high
myopia of -7.0 dioptres, which was complicated by
cataract in the right eye at age 32 years. The variants
were distributed across the LMX1B gene. Three
affected families exhibited intragenic variants: two
nonsense variants and one missense variant. Among
these, two were novel variants, whereas the LMX1B
c.745C>T nonsense variant had been previously
reported.2 3 Three individuals from two unrelated
families harboured novel exon deletions. The
clinical and molecular findings of these patients are
summarised in the Figure, the online supplementary Figure and the online supplementary Table.
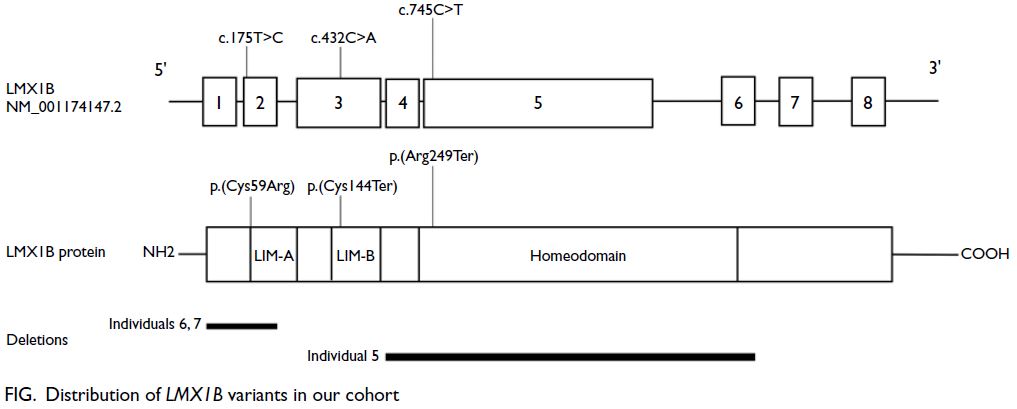
Figure. Distribution of LMX1B variants in our cohort
Nail-patella syndrome is a rare condition
characterised by substantial inter- and intrafamilial
variability. Some individuals in our cohort
(Individuals 2, 3, and 5) showed symptoms from early
infancy, whereas others (Individuals 4 and 7) were
diagnosed in adulthood during incidental family
screening or routine health checks. Nail anomalies
are the most common feature, identified in >95% of
affected individuals, consistent with observations
in our cohort.1 4 In particular, Individuals 1 and
2 had triangular lunulae, a feature considered
pathognomonic for NPS. Skeletal anomalies
involving the knee and elbow are described in
70% of affected individuals.1 4 The detection of
hypoplastic patellae may be challenging through
physical examination alone, but iliac horns are often
evident on radiographs. Although the proportion
of knee involvement in our cohort aligns with
previous literature reports, elbow involvement was
considerably less frequent.1 4
The majority of individuals with NPS inherit
pathogenic variants from a parent—an estimated
15% of cases in the literature have been attributed
to de novo variants.1 All individuals in our cohort
had a positive family history except for Individual 5.
Ascertainment bias is a concern because some at-risk
relatives with mild symptoms, often comprising isolated nail dystrophy, declined molecular
confirmation. Consequently, the clinical burden
of NPS may be overestimated, but underdiagnosis
remains prevalent. Renal involvement, reported in
up to half of affected individuals and associated with
the risk of end-stage kidney disease,1 5 was identified
in only one individual in our cohort (Individual
1), who displayed microscopic haematuria during
surveillance. No individuals in our cohort showed
renal impairment, which may be explained by the
age-dependent penetrance of renal symptoms and
the relatively young age of these individuals. Other
rare features previously reported in association with
NPS, such as vascular anomalies and reduced bone
mineral density, were not observed in our cohort.
Thus far, LMX1B is the only gene implicated in
NPS; it is typically interrogated through sequencing
analysis, which detects up to 85% of variants,
followed by dosage analysis using techniques such as
multiplex ligation-dependent probe amplification.1
Although most pathogenic variants (80%) reportedly
occur in the LIM domain of LMX1B,1 the variants
identified in our cohort were distributed throughout
the gene, with only c.175T>C and c.432C>A located
in the LIM-A and LIM-B domains, respectively
(Figure). Two families in our cohort harboured exon
deletions. The mutation spectrum observed in our
cohort does not entirely align with descriptions in
the literature, but this discrepancy may reflect the
relatively small number of patients included in the
study. Ophthalmic involvement has been reported in
10% to 25% of affected individuals.1 4 In our cohort,
two affected individuals from the same family
(Individuals 2 and 3), carrying the c.745C>T variant,
exhibited severe ocular manifestations and impaired
vision from early infancy. While this variant has
been described in the literature, it was not previously
linked to ophthalmic involvement in NPS.2 3 Further research is required to determine whether ethnicity
influences phenotypic and genotypic differences,
considering that our cohort mostly comprised
individuals of Chinese ethnicity.
Author contributions
Concept or design: LT Leung, SKL Ho.
Acquisition of data: LT Leung, SKL Ho, SSW Cheng, IFM Lo, HM Luk.
Analysis or interpretation of data: LT Leung, SKL Ho, WC Yiu.
Drafting of the manuscript: LT Leung, SKL Ho.
Critical revision of the manuscript for important intellectual content: SSW Cheng, IFM Lo, HM Luk.
Acquisition of data: LT Leung, SKL Ho, SSW Cheng, IFM Lo, HM Luk.
Analysis or interpretation of data: LT Leung, SKL Ho, WC Yiu.
Drafting of the manuscript: LT Leung, SKL Ho.
Critical revision of the manuscript for important intellectual content: SSW Cheng, IFM Lo, HM Luk.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
The authors declared no conflicts of interest.
Funding/support
This commentary received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patients were treated in accordance with the Declaration
of Helsinki and provided informed consent for publication
of this commentary, including the publication of clinical
photos.
Supplementary material
The supplementary material was provided by the authors, and
some information may not have been peer reviewed. Accepted
supplementary material will be published as submitted by the
authors, without any editing or formatting. Any opinions
or recommendations discussed are solely those of the
author(s) and are not endorsed by the Hong Kong Academy of Medicine and the Hong Kong Medical Association.
The Hong Kong Academy of Medicine and the Hong Kong
Medical Association disclaim all liability and responsibility
arising from any reliance placed on the content.
References
1. Sweeney E, Hoover-Fong JE, McIntosh I, et al. Nail-Patella
Syndrome. In GeneReviews® [Internet]. Seattle (WA):
University of Washington, Seattle; 1993.
2. Dunston JA, Hamlington JD, Zaveri J, et al. The human LMX1B gene: transcription unit, promoter, and pathogenic mutations. Genomics 2004;84:565-76.Crossref
3. Tayebi N, Charng WL, Dickson PI, Dobbs MB, Gurnett CA.
Diagnostic yield of exome sequencing in congenital vertical
talus. Eur J Med Genet 2022;65:104514. Crossref
4. Ghoumid J, Petit F, Holder-Espinasse M, et al. Nail-patella
syndrome: clinical and molecular data in 55 families raising
the hypothesis of a genetic heterogeneity. Eur J Hum Genet
2016;24:44-50. Crossref
5. Tigchelaar S, Lenting A, Bongers EM, van Kampen A. Nail-patella
syndrome: knee symptoms and surgical outcomes.
A questionnaire-based survey. Orthop Traumatol Surg Res
2015;101:959-62. Crossref