Hong Kong Med J 2025;31:Epub 12 Feb 2025
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Prevalence, risk factors, and outcomes of systemic
sclerosis–associated interstitial lung disease in a Chinese population
Dennis TH Chan, MRCP (UK), FHKAM (Medicine)1; Lydia HP Tam, MRCP (UK), FHKAM (Medicine)2; Tommy TO Lam, MRCP (UK), FHKAM (Medicine)2; Jacqueline So, MRCP (UK), FHKAM (Medicine)2; LY Ho, MRCP (UK), FHKAM (Medicine)2; LS Tam, MRCP (UK), FRCP (Lond)2; Ho So, FHKAM (Medicine), FRCP (Lond)2
1 Department of Medicine, Alice Ho Miu Ling Nethersole Hospital, Hong Kong SAR, China
2 Department of Medicine and Therapeutics, Prince of Wales Hospital, The Chinese University of Hong Kong, Hong Kong SAR, China
3 Department of Medicine and Geriatrics, Tai Po Hospital, Hong Kong
Corresponding author: Dr Dennis TH Chan (cdt978@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Abstract
Introduction: Systemic sclerosis–associated
interstitial lung disease (SSc-ILD) is a leading cause
of mortality among systemic sclerosis (SSc) patients.
This multicentre cohort study sought to determine
the prevalence of SSc-ILD, identify risk factors for
ILD development in SSc patients, and explore poor
prognostic factors in SSc-ILD patients.
Methods: Medical records were retrospectively
reviewed for Chinese patients who met the 2013
American College of Rheumatology/European
League Against Rheumatism classification criteria
for SSc. Univariable and multivariable analyses
were performed to compare SSc patients with and
without ILD, as well as SSc-ILD patients with and
without disease progression. Survival analysis was
also conducted.
Results: The study cohort comprised 223 SSc patients
with a median follow-up duration of 8.1 years. The
prevalence of ILD was 49.8%. A history of bibasal
crackles (hazard ratio [HR]=2.813; P=0.001) was
independently associated with ILD development.
Among ILD patients, 64.1% exhibited progressive
disease. An elevated C-reactive protein (CRP) level
at ILD diagnosis (HR=1.064; P=0.002) constituted
an independent predictor of ILD progression. The
overall mortality rate was 24.2% and pneumonia was the most common cause of death. Predictors of
mortality included age at SSc diagnosis (HR=1.101;
P=0.002), history of smoking (HR=5.173; P=0.028),
and CRP level at SSc diagnosis (HR=1.103; P=0.009).
Conclusion: Interstitial lung disease was prevalent
among SSc patients in this cohort and the majority
exhibited disease progression. Comprehensive
clinical assessment, supported by investigations such
as CRP level measurement, is essential to identify
predictors of poor prognosis.
New knowledge added by this study
- Interstitial lung disease (ILD) is common and often progressive among systemic sclerosis (SSc) patients in the Hong Kong Chinese population.
- Baseline C-reactive protein level is independently associated with ILD progression and mortality in SSc patients.
- Interstitial lung disease screening is recommended for all SSc patients.
- C-reactive protein level may serve as a predictor of ILD progression and mortality in SSc patients.
- Prospective studies are necessary to develop personalised monitoring and treatment strategies.
Introduction
Systemic sclerosis (SSc) is a heterogeneous
connective tissue disorder involving multiple organ
systems. Its subtypes comprise limited cutaneous
SSc (lcSSc) and diffuse cutaneous SSc (dcSSc).1
Common features include Raynaud’s phenomenon,
skin sclerosis, and musculoskeletal inflammation.
Organ-based manifestations, such as interstitial lung
disease (ILD), pulmonary hypertension (PH), and scleroderma renal crisis, are particularly important
because they substantially affect patient quality
of life and survival. Systemic sclerosis–associated
interstitial lung disease (SSc-ILD) is the leading
cause of mortality in SSc, contributing to 35% of
disease-related deaths.2 In Hong Kong, SSc has one
of the highest standardised mortality ratios among
rheumatic diseases.3
Systemic sclerosis–associated interstitial lung disease arises from chronic microinjuries to lung
endothelial and epithelial cells, which activate the
immune system and lead to the recruitment and
transformation of fibroblasts into myofibroblasts that
secrete excessive collagen-rich extracellular matrix.4 5
This pathological process causes pathological lung
stiffness and architectural disruption, producing
restrictive lung disease through reductions of lung
compliance and volume.6
It is well established that there is an ethnic
disparity in SSc-ILD; prevalence rates considerably
vary among ethnic groups, ranging from 25% to
90%.7 The prevalence of SSc-ILD is reportedly higher
in Asian populations than in Western populations.8
However, data concerning the prevalence and
predictive factors of SSc-ILD in Southern Chinese
individuals remain limited. A prospective case-control
study investigating functioning and health-related
quality of life in Hong Kong showed that
among 78 SSc patients recruited, 24 (30.8%) had
ILD.9
The clinical course of SSc-ILD ranges from
asymptomatic presentation to rapidly progressive
disease, which can lead to mortality. Severe disease
develops in approximately 25% to 33% of SSc-ILD
patients.4 Thus, it is essential to identify patients with
early-stage SSc who are asymptomatic but exhibit
a risk of ILD development and progression. This approach enables closer monitoring and facilitates
timely treatment. Numerous risk factors for ILD
development and progression in SSc patients have
been reported.8 10 11 According to the 2020 European
consensus statements on the identification and
management of ILD in SSc,10 predictive factors
include respiratory symptoms, smoking history,
ethnicity (eg, native American or African heritage),
dcSSc, presence of anti-topoisomerase antibody
(ATA), and male sex. However, most of these
findings were based on studies conducted in Western
populations.10
To improve the identification and management
of SSc patients at risk of ILD development or
progression, we conducted a multicentre study
that aimed to assess the prevalence of SSc-ILD in
the Hong Kong Chinese population, investigate
associated risk factors, and identify potential
indicators of poor prognosis. The findings of this
study are expected to enhance early detection and
monitoring of ILD in SSc patients, enabling timely
and effective interventions.
Methods
Study design and patients
This retrospective longitudinal study included SSc
patients who attended Alice Ho Miu Ling Nethersole
Hospital, Prince of Wales Hospital, and North
District Hospital. These patients were identified via
the Clinical Data Analysis and Reporting System, a
database established for record keeping and research
purposes in Hong Kong, which has been utilised
in epidemiological studies.12 The International
Classification of Diseases, Ninth Revision, Clinical
Modification code 710.1 (Systemic sclerosis) was
used to identify SSc patients within the Clinical
Data Analysis and Reporting System. The search
period spanned from January 2008 to December
2022. Clinical information for each patient was
reviewed in the electronic health record. Patients
were included if they had attended more than one
follow-up appointment and met the 2013 American
College of Rheumatology/European League Against
Rheumatism classification criteria for SSc.13
Exclusion criteria were age at onset <18 years, overlap
syndrome, and non-Chinese ethnicity. Patients
with ILD were identified based on radiologists’
reports of high-resolution computed tomography
(HRCT) of the thorax. For patients without HRCT
records, chest radiographs were reviewed to identify
evidence of ILD. Investigations, treatments, and the
frequency of follow-ups were determined by the
treating physicians.
Clinical variable collection
Demographic variables, including sex, smoking history, age at symptom onset, age at SSc diagnosis, and age at ILD diagnosis, were recorded. The first
clinical symptoms attributed to SSc, as judged by the
treating physicians, and symptoms observed during
the follow-up period were documented. These
symptoms included Raynaud’s phenomenon, puffy
fingers, sclerodactyly, digital ulcers, oesophageal
dysmotility, arthralgia, dyspnoea, and cough.14 The
presence of bibasal crackles on physical examination
by the treating physicians was also documented. The
status of PH was recorded based on findings from
echocardiography or right heart catheterisation.
Disease duration was defined as the time from onset
of the first symptom to the last visit. The SSc subtype
was categorised as dcSSc or lcSSc based on the
extent of skin involvement, using criteria established
by LeRoy and Medsger.1
Laboratory data, including autoantibodies,
C-reactive protein (CRP), and erythrocyte
sedimentation rate (ESR) levels, were recorded.
C-reactive protein and ESR levels at baseline and
at ILD diagnosis were documented. Pulmonary
function test (PFT) results at baseline and at the
latest available assessment were retrieved. Forced
expiratory volume in 1 second, forced vital capacity
(FVC), and diffusing capacity of the lungs for carbon
monoxide (DLCO) were recorded. In ILD cases,
the radiological pattern on HRCT, including non-specific
interstitial pneumonitis, usual interstitial
pneumonia, or other patterns, was noted.
Systemic sclerosis–associated interstitial
lung disease outcomes were assessed based on ILD
progression and mortality. Disease progression was
defined as an increase in ILD extent on serial HRCT,
as reported by radiologists, or a decline in FVC of
≥10% from baseline. Alternatively, progression was
defined as an FVC decline of 5% to 9% accompanied
by a DLCO decline of ≥15%.15 Causes of death were
categorised as SSc-related or SSc-unrelated, based
on assessment by the treating physicians (when
available) or the authors. Clinical variables with
>20% missing data were excluded from statistical
analyses.
Statistical analyses
Descriptive data for continuous variables were
presented as mean±standard deviation or median
(interquartile range [IQR]), as appropriate.
Categorical variables were presented as numbers
with percentages. Student’s t test or the Mann-Whitney U test was used for comparisons of
continuous variables, depending on the data
distribution. Categorical variables were compared
using Fisher’s exact test or the Chi squared test.
Patients with and without ILD were compared
using univariable and multivariable Cox regression
analyses to identify risk factors associated with the
development of SSc-ILD. Among SSc-ILD patients,
those displaying progressive ILD were compared with those lacking progression via univariable
and multivariable analyses to identify risk factors
for disease progression. The univariate effects of
covariates on survival were evaluated using Kaplan–Meier curves; the log-rank test was utilised to assess
differences in survival. Multivariable Cox regression
analyses were conducted to identify independent
predictors of adverse outcomes. Variables with
P value <0.2 in univariable analyses were included
in the multivariable Cox regression analysis. All
statistical analyses were performed using SPSS
(Windows version 27.0; IBM Corp, Armonk [NY],
United States). P values <0.05 were considered
statistically significant.
Results
Demographics and clinical characteristics
In total, 223 SSc patients were included in this study
(Fig). Table 1 summarises the patients’ baseline
characteristics. The median follow-up duration was
8.1 years (IQR=4.0-10.2) and the total cumulative
follow-up period was 1951 person-years. The
majority of patients were female (86.1%). The median
age at SSc diagnosis was 55 years (IQR=48-64). A
majority of patients (86.5%) underwent HRCT scans
during the follow-up period. Among those without
HRCT, none had chest radiographs suggestive of
ILD. Limited cutaneous SSc was the most common
subtype, displayed by 71.3% of the cohort. Anti-topoisomerase
antibody was the most frequently
detected autoantibody, present in 39.0% of patients.
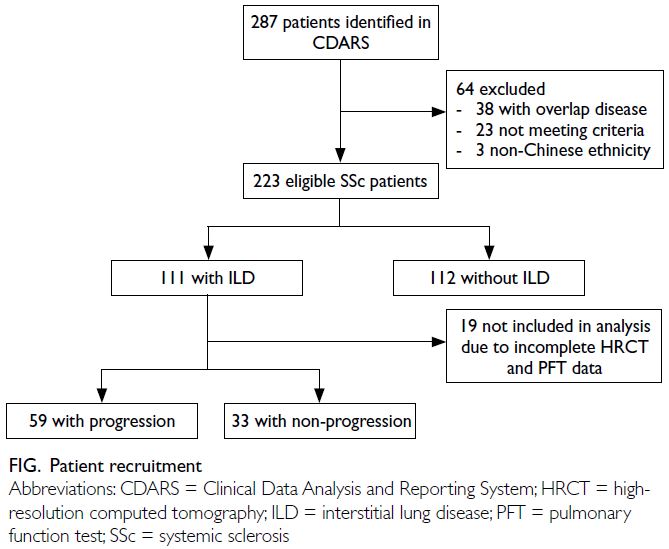
Figure. Patient recruitment
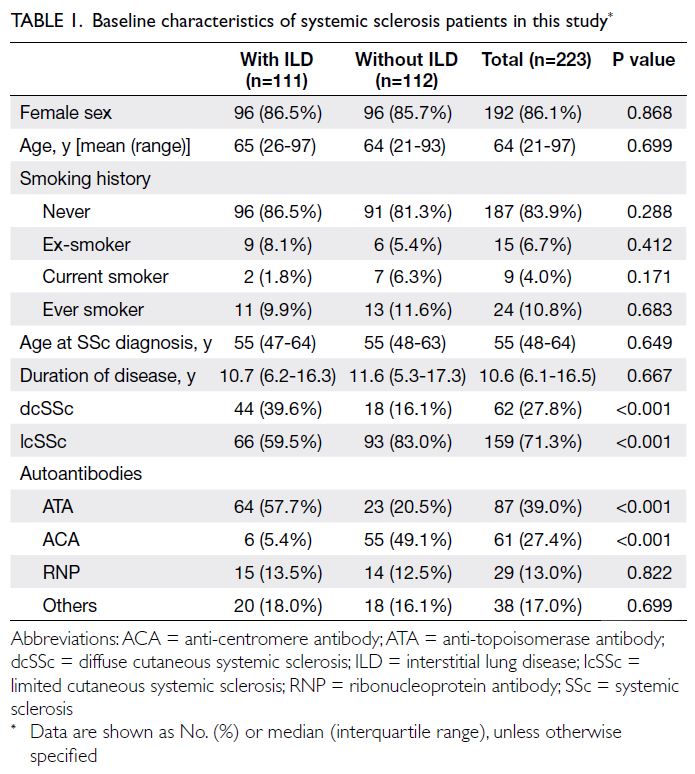
Table 1. Baseline characteristics of systemic sclerosis patients in this study
The overall prevalence of ILD among SSc
patients was 49.8%. The age at ILD diagnosis ranged from 20 to 85 years, with a median of 57 years. Most
patients in the SSc-ILD subgroup were female (86.5%)
and non-smokers (86.5%); these characteristics did
not significantly differ relative to patients without
ILD (Table 1). The median interval from onset of
the first SSc symptom to ILD diagnosis was 2.4
years (IQR=1.3-5.4). Among ILD cases, 51.3% were
diagnosed within the first 3 years after SSc symptom
onset, and 64.0% were diagnosed within 5 years. Of
the ILD patients, 18.9% were asymptomatic, whereas
symptomatic patients experienced a median interval
of 2.4 years (IQR=1.2-6.3) from respiratory symptom
onset to ILD diagnosis.
The frequency of dcSSc was significantly
greater in patients with ILD than in patients without
ILD (39.6% vs 16.1%; P<0.001). Conversely, lcSSc
was more common in patients without ILD than in
patients with ILD (83.0% vs 59.5%; P<0.001). In the
ILD group, ATA was the most frequently detected
autoantibody (57.7%), whereas anti-centromere
antibody (ACA) was more common in the non-ILD
group (49.1%) [Table 1].
The frequencies of non-respiratory clinical
features were comparable between the ILD and
non-ILD groups. However, respiratory features,
including dyspnoea, cough and bibasal crackles
significantly differed between the two groups, both
at presentation and during follow-up (P<0.001 for all comparisons). Pulmonary hypertension
was significantly more frequent in the ILD group
throughout the follow-up period (19.8% vs
2.7%; P<0.001). The ILD group also exhibited a
numerically higher baseline ESR, with a median of
21.5 mm/hr (IQR=14-40.5), whereas the non-ILD
group displayed a median of 18 mm/hr (IQR=11-30;
P=0.074) [online supplementary Table 1].
Associative factors of interstitial lung disease
development
Univariable analysis showed that several factors
were associated with the presence of ILD (online supplementary Table 2). These included dcSSc,
ATA, history of dyspnoea, history of cough, history
of bibasal crackles, history of PH, and baseline ESR
level. Conversely, ACA and lcSSc were negatively
associated with ILD development. According to
multivariable Cox regression analysis, a history of
bibasal crackles was independently associated with
the presence of ILD, and a history of dyspnoea
showed a trend towards significance.
Predictors of interstitial lung disease
progression
Among patients with ILD, 64.1% exhibited
progression during follow-up. Patients with
progressive ILD were younger at ILD diagnosis,
displaying a mean age of 54 years (range, 20-85)
compared with 60 years (range, 31-81; P=0.051) in
patients with non-progressive ILD. The proportions
of dcSSc and lcSSc were similar between the
progressive and non-progressive ILD groups. Anti-topoisomerase
antibody was the predominant
autoantibody in both groups, with proportions of
62.7% and 54.5%, respectively (P=0.444) [online supplementary Table 3]. Regarding clinical
characteristics, only a history of digital ulcers
showed a significant difference; its prevalence was
higher in the progressive ILD group (42.4% vs 15.2%;
P=0.008) [online supplementary Table 4].
Table 2 compares the results of laboratory and
PFT between the progressive and non-progressive
ILD groups. C-reactive protein levels at both SSc
diagnosis and ILD diagnosis were higher in the
progressive ILD group; however, only CRP level at
ILD diagnosis showed a trend towards significance
(P=0.130). The latest values for the predicted
percentages of forced expiratory volume in 1 second,
FVC and DLCO were significantly lower in the
progressive ILD group (all P≤0.001), but baseline
values did not differ between the groups. Regarding
HRCT patterns, no significant differences were
observed between the two groups.
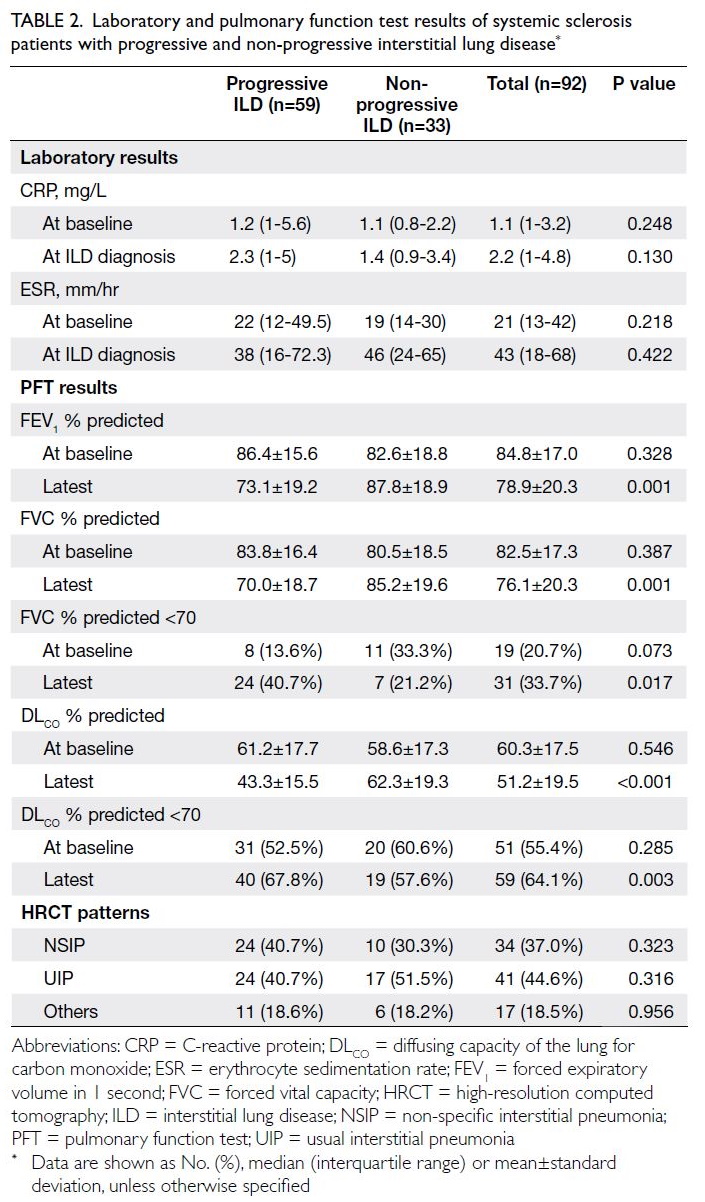
Table 2. Laboratory and pulmonary function test results of systemic sclerosis patients with progressive and non-progressive interstitial lung disease
The results of the Cox regression analysis for
ILD progression are presented in Table 3. In the
univariable analysis, factors associated with ILD
progression included CRP level at ILD diagnosis (hazard ratio [HR]=1.504; P=0.005) and the latest
predicted percentage of DLCO (HR=0.962; P<0.001).
Multivariable analysis identified CRP level at ILD
diagnosis (HR=1.064; P=0.002) as an independent
factor associated with ILD progression, whereas a
history of digital ulcers (HR=1.874; P=0.076) showed
a trend towards significance.
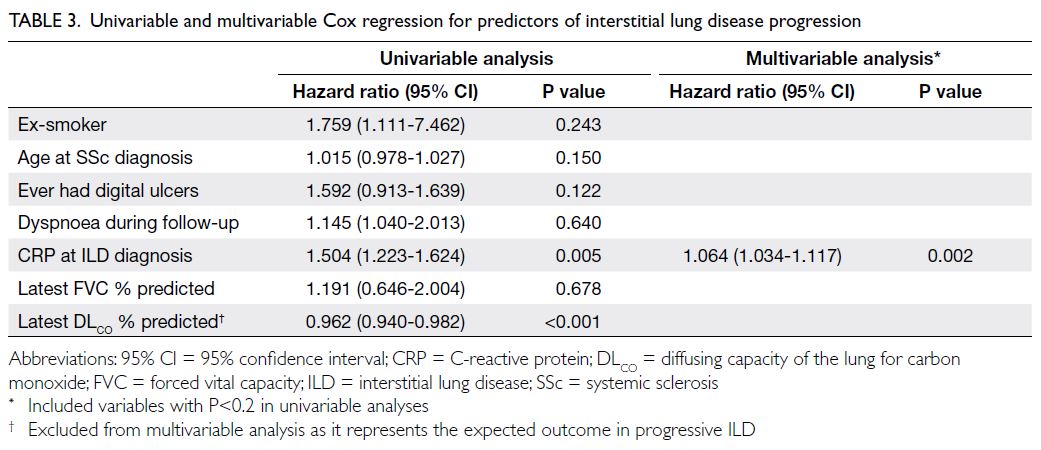
Table 3. Univariable and multivariable Cox regression for predictors of interstitial lung disease progression
Mortality
The overall mortality rate in the cohort during
the follow-up period was 24.2%; a higher rate
was observed in the SSc-ILD group relative to
the non-ILD group (29.7% vs 18.8%, P=0.056)
[online supplementary Fig]. Among the causes of
death, infections were most common, followed
by malignancy (online supplementary Table 5). In
patients with ILD, 63.6% of deaths resulted from
pneumonia; this proportion was 42.9% among
patients without ILD. The univariable analysis
indicated that factors associated with mortality
were older age at SSc diagnosis, male sex, history of
smoking, presence of PH, baseline CRP level, and
baseline ESR level. Multivariable analysis revealed
the following independent predictors of mortality:
older age at SSc diagnosis (HR=1.101; P=0.002),
history of smoking (HR=5.173; P=0.028), and higher
baseline CRP level (HR=1.103; P=0.009) [Table 4].

Table 4. Univariable and multivariable Cox regression for predictors of mortality
Discussion
We observed an SSc-ILD prevalence of 49.8% in a multicentre cohort of Southern Chinese SSc patients.
Among Asian countries, the reported prevalence
of SSc-ILD varies. In Korea, prevalence rates range
from 40% to 58%,16 17 whereas in Japan, they range
from 42% to 51%.18 19 However, considerably higher
prevalence estimates of 63% to 85% have been
reported in centres from Northern China.20 21 These
findings suggest ethnic or geographic variations
in the prevalence of SSc-ILD within the Asian
population. Given the high prevalence in our
cohort and the observed delay between respiratory
symptom onset and ILD diagnosis (median=2.4
years), early universal screening for ILD is necessary
among SSc patients. This is particularly important
because a substantial proportion of patients (18.9%)
were asymptomatic.
Consistent with previous studies, our findings
confirmed that in Chinese SSc patients, the dcSSc
subtype and presence of ATA were associated with
a higher likelihood of ILD development, whereas the
lcSSc subtype and presence of ACA were inversely
related to ILD risk.11 22 23 Also, our study showed
that a history of bibasal crackles was independently
associated with ILD development, similar to the
findings of a retrospective cohort study in South
Africa.24 However, it is important to recognise that
the presence of crackles often reflects established
disease, and the new onset of respiratory symptoms may indicate ILD development. Irrespective of
the presence of respiratory symptoms, all SSc
patients are recommended to undergo screening
for ILD via HRCT and PFTs, as specified by expert
consensuses.10 25 Regular auscultation for bibasal
crackles during follow-up is equally important
because it facilitates the identification of individuals
who may require repeat investigations.
C-reactive protein has been proposed as a
biomarker for predicting SSc-ILD progression.26
Similar to our findings, a retrospective cohort
study in France27 revealed a significant difference in CRP levels between SSc patients with and without
ILD (P=0.003). The multivariate analysis in that
study also demonstrated a negative correlation
between CRP levels and FVC.27 C-reactive protein
production is driven by interleukin 6, and interleukin
6 inhibitors have shown efficacy in preserving lung
function among SSc-ILD patients during a phase
three randomised controlled trial.28 These findings
provide a mechanistic rationale for using CRP levels
to identify SSc-ILD patients who may benefit from
early investigation and treatment.
The cumulative survival rates reported in
our study align with those observed in Western
populations.11 29 However, a European Scleroderma
Trials and Research Group cohort study conducted
in China20 demonstrated a higher cumulative survival rate of 87.8% at 10 years, a lower overall
mortality rate of 8.9%, and fewer SSc-ILD–related
deaths (2.5%). This disparity may be attributed to
the higher frequency of infection-related deaths and
the greater proportion of patients with progressive
disease in our cohort. Although assessments of
treatment regimen and response were beyond
the scope of our study, due to the confounding by
indication involved in its retrospective design,
immunosuppressive agents commonly used in the
past may have predisposed patients to infections.
Indeed, infection has previously been identified as
the leading cause of death in local SSc patients.3
Considering the high rate of infection-related
mortality, recently available antifibrotic treatments
may be preferable to immunosuppressive therapy in selected patients who exhibit increased infection
risk. Furthermore, consistent with well-established
evidence, we identified increased age15 30 31 32 and
elevated CRP levels at SSc diagnosis33 34 as predictors
of mortality. It remains unclear whether more
aggressive early treatment in patients with elevated
baseline CRP levels would improve survival; further
investigation is warranted.
Limitations
Some limitations should be acknowledged in our
study. The data were extracted from the electronic
health record, making undercoding of diagnoses
unavoidable. Due to the retrospective study design,
some clinical data essential to this study might
not have been fully documented, and disease
progression monitoring was not systematic, which
could introduce bias. The presence of symptoms
and ILD was assessed by the treating physicians
and radiologists, respectively; these assessments
potentially lacked specificity or sensitivity. Follow-up
investigations were primarily ordered based on
clinical judgement, leading to potential selection
bias. Our analyses did not adjust for patients
with progressive disease who may have received
treatment leading to ILD stabilisation, which could
have resulted in classification of their ILD as non-progressive.
Furthermore, no standardised criteria
currently exist for defining SSc-ILD progression.
Quantitative assessments of ILD involvement on
HRCT, such as percentage involvement or the
Warrick score,35 and the extensiveness of skin
disease using the modified Rodnan skin score,36 were
also unavailable.
Conclusion
This is the first multicentre cohort study to
investigate SSc-ILD in Hong Kong. Our findings
demonstrated a high prevalence of ILD among
Chinese SSc patients, with a significant proportion
of these patients exhibiting disease progression.
Universal ILD screening is recommended for SSc
patients, with particular attention to those who
develop respiratory symptoms and signs. In addition
to imaging and PFTs, CRP levels could serve as a
biomarker for ILD progression and poor prognosis.
Author contributions
Concept or design: DTH Chan, H So.
Acquisition of data: DTH Chan, LHP Tam.
Analysis or interpretation of data: DTH Chan, H So.
Drafting of the manuscript: DTH Chan, H So.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: DTH Chan, LHP Tam.
Analysis or interpretation of data: DTH Chan, H So.
Drafting of the manuscript: DTH Chan, H So.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Declaration
The results of this study were presented as poster presentation
at 26th Asia-Pacific League of Associations for Rheumatology
Congress 2024 in Singapore, 21-25 August 2024.
Funding/support
This research received no specific grant from any funding
agency in the public, commercial, or not-for-profit sectors.
Ethics approval
This research was approved by the Joint Chinese University of
Hong Kong–New Territories East Cluster Clinical Research
Ethics Committee, Hong Kong (Ref No.: CREC-2023-393).
The requirement for informed patient consent was waived by
the Committee due to the retrospective nature of the research.
Supplementary material
The supplementary material was provided by the authors, and
some information may not have been peer reviewed. Accepted
supplementary material will be published as submitted by the
authors, without any editing or formatting. Any opinions
or recommendations discussed are solely those of the
author(s) and are not endorsed by the Hong Kong Academy
of Medicine and the Hong Kong Medical Association.
The Hong Kong Academy of Medicine and the Hong Kong
Medical Association disclaim all liability and responsibility
arising from any reliance placed on the content.
References
1. LeRoy EC, Medsger TA Jr. Criteria for the classification of
early systemic sclerosis. J Rheumatol 2001;28:1573-6.
2. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk
factors for death in systemic sclerosis: a study from the
EULAR Scleroderma Trials and Research (EUSTAR)
database. Ann Rheum Dis 2010;69:1809-15. Crossref
3. Mok CC, Kwok CL, Ho LY, Chan PT, Yip SF. Life expectancy,
standardized mortality ratios, and causes of death in six
rheumatic diseases in Hong Kong, China. Arthritis Rheum
2011;63:1182-9. Crossref
4. Khanna D, Tashkin DP, Denton CP, Renzoni EA, Desai SR,
Varga J. Etiology, risk factors, and biomarkers in systemic
sclerosis with interstitial lung disease. Am J Respir Crit
Care Med 2020;201:650-60. Crossref
5. Mostmans Y, Cutolo M, Giddelo C, et al. The role of
endothelial cells in the vasculopathy of systemic sclerosis: a
systematic review. Autoimmun Rev 2017;16:774-86. Crossref
6. Khanna D, Lescoat A, Roofeh D, et al. Systemic sclerosis–associated interstitial lung disease: how to incorporate two
Food and Drug Administration–approved therapies in
clinical practice. Arthritis Rheumatol 2022;74:13-27. Crossref
7. Qiu M, Nian X, Pang L, Yu P, Zou S. Prevalence and risk
factors of systemic sclerosis–associated interstitial lung
disease in East Asia: a systematic review and meta-analysis.
Int J Rheum Dis 2021;24:1449-59. Crossref
8. Chan DT, So H. Systemic sclerosis–associated interstitial
lung disease: prevalence and risk factors. J Clin Rheumatol Immunol 2023;23:15-24. Crossref
9. Chan PT, Mok CC, Chan KL, Ho LY. Functioning and
health-related quality of life in Chinese patients with
systemic sclerosis: a case-control study. Clin Rheumatol
2014;33:659-66. Crossref
10. Hoffmann-Vold AM, Maher TM, Philpot EE, et al. The
identification and management of interstitial lung disease
in systemic sclerosis: evidence-based European consensus
statements. Lancet Rheumatol 2020;2:e71-83. Crossref
11. Nihtyanova SI, Schreiber BE, Ong VH, et al. Prediction
of pulmonary complications and long-term survival in
systemic sclerosis. Arthritis Rheumatol 2014;66:1625-35. Crossref
12. So H, So J, Lam TT, et al. Performance of the 2017
European Alliance of Associations for Rheumatology/American College of Rheumatology classification criteria
in patients with idiopathic inflammatory myopathy and
anti–melanoma differentiation–associated protein 5
positivity. Arthritis Rheumatol 2022;74:1588-92. Crossref
13. van den Hoogen F, Khanna D, Fransen J, et al. 2013
classification criteria for systemic sclerosis: an American
College of Rheumatology/European League against
Rheumatism Collaborative Initiative. Arthritis Rheum
2013;65:2737-47. Crossref
14. van den Hombergh WM, Carreira PE, Knaapen-Hans HK,
van den Hoogen FH, Fransen J, Vonk MC. An easy
prediction rule for diffuse cutaneous systemic sclerosis
using only the timing and type of first symptoms and
auto-antibodies: derivation and validation. Rheumatology
(Oxford) 2016;55:2023-32. Crossref
15. Goh NS, Hoyles RK, Denton CP, et al. Short-term
pulmonary function trends are predictive of mortality in
interstitial lung disease associated with systemic sclerosis.
Arthritis Rheumatol 2017;69:1670-8. Crossref
16. Jung E, Suh CH, Kim HA, Jung JY. Clinical characteristics
of systemic sclerosis with interstitial lung disease. Arch
Rheumatol 2018;33:322-7. Crossref
17. Kim J, Park SK, Moon KW, et al. The prognostic factors
of systemic sclerosis for survival among Koreans. Clin
Rheumatol 2010;29:297-302. Crossref
18. Sekiguchi A, Inoue Y, Yamazaki S, et al. Prevalence and
clinical characteristics of earlobe crease in systemic
sclerosis: possible association with vascular dysfunction. J
Dermatol 2020;47:870-5. Crossref
19. Aozasa N, Hatano M, Saigusa R, et al. Clinical significance
of endothelial vasodilatory function evaluated by EndoPAT
in patients with systemic sclerosis. J Dermatol 2020;47:609-14. Crossref
20. Hu S, Hou Y, Wang Q, Li M, Xu D, Zeng X. Prognostic
profile of systemic sclerosis: analysis of the clinical
EUSTAR cohort in China. Arthritis Res Ther 2018;20:235. Crossref
21. Wang J, Assassi S, Guo G, et al. Clinical and serological
features of systemic sclerosis in a Chinese cohort. Clin
Rheumatol 2013;32:617-21. Crossref
22. Sánchez-Cano D, Ortego-Centeno N, Callejas JL, et al.
Interstitial lung disease in systemic sclerosis: data from the Spanish scleroderma study group. Rheumatol Int
2018;38:363-74. Crossref
23. Gelber AC, Manno RL, Shah AA, et al. Race and association
with disease manifestations and mortality in scleroderma:
a 20-year experience at the Johns Hopkins Scleroderma
Center and review of the literature. Medicine (Baltimore)
2013;92:191-205. Crossref
24. Ashmore P, Tikly M, Wong M, Ickinger C. Interstitial
lung disease in South Africans with systemic sclerosis.
Rheumatol Int 2018;38:657-62. Crossref
25. Rahaghi FF, Hsu VM, Kaner RJ, et al. Expert consensus
on the management of systemic sclerosis–associated
interstitial lung disease. Respir Res 2023;24:6. Crossref
26. Distler O, Assassi S, Cottin V, et al. Predictors of progression
in systemic sclerosis patients with interstitial lung disease.
Eur Respir J 2020;55:1902026. Crossref
27. Chikhoune L, Brousseau T, Morell-Dubois S, et al.
Association between routine laboratory parameters and
the severity and progression of systemic sclerosis. J Clin
Med 2022;11:5087. Crossref
28. Khanna D, Lin CJ, Furst DE, et al. Long-term safety
and efficacy of tocilizumab in early systemic sclerosis–interstitial lung disease: open-label extension of a phase 3
randomized controlled trial. Am J Respir Crit Care Med
2022;205:674-84. Crossref
29. Pokeerbux MR, Giovannelli J, Dauchet L, et al. Survival
and prognosis factors in systemic sclerosis: data of a French
multicenter cohort, systematic review, and meta-analysis
of the literature. Arthritis Res Ther 2019;21:86. Crossref
30. Volkmann ER, Tashkin DP, Sim M, et al. Short-term
progression of interstitial lung disease in systemic sclerosis
predicts long-term survival in two independent clinical
trial cohorts. Ann Rheum Dis 2019;78:122-30. Crossref
31. Volkmann ER, Saggar R, Khanna D, et al. Improved
transplant-free survival in patients with systemic sclerosis–associated pulmonary hypertension and interstitial lung
disease. Arthritis Rheumatol 2014;66:1900-8. Crossref
32. Takei R, Arita M, Kumagai S, et al. Radiographic fibrosis
score predicts survival in systemic sclerosis–associated
interstitial lung disease. Respirology 2018;23:385-91. Crossref
33. Liu X, Mayes MD, Pedroza C, et al. Does C-reactive
protein predict the long-term progression of interstitial
lung disease and survival in patients with early systemic
sclerosis? Arthritis Care Res (Hoboken) 2013;65:1375-80. Crossref
34. Le Gouellec N, Duhamel A, Perez T, et al. Predictors
of lung function test severity and outcome in systemic
sclerosis–associated interstitial lung disease. PLoS One
2017;12:e0181692. Crossref
35. Warrick JH, Bhalla M, Schabel SI, Silver RM. High
resolution computed tomography in early scleroderma
lung disease. J Rheumatol 1991;18:1520-8.
36. Khanna D, Furst DE, Clements PJ, et al. Standardization of
the modified Rodnan skin score for use in clinical trials of
systemic sclerosis. J Scleroderma Relat Disord 2017;2:11-8. Crossref