Hong Kong Med J 2024 Aug;30(4):328–30 | Epub 29 May 2024
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Long-surviving Neu-Laxova syndrome confirmed by whole exome sequencing: a case report
CW Kong, MSc, FHKAM (Obstetrics and Gynaecology)1; YY Li, MSc, FHKAM (Obstetrics and Gynaecology)1; Sandy LK Au, BSc, PhD2; Anita SY Kan, MPH, FHKAM (Obstetrics and Gynaecology)3; Martin MC Chui, BSc4; Brian HY Chung, MD, FHKAM (Paediatrics)5; YC Ho, MRCP, FHKAM (Paediatrics)6; William WK To, MD, FRCOG1
1 Department of Obstetrics and Gynaecology, United Christian Hospital, Hong Kong SAR, China
2 Department of Obstetrics and Gynaecology, School of Clinical Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, China
3 Department of Obstetrics and Gynaecology, Queen Mary Hospital, Hong Kong SAR, China
4 Department of Paediatrics and Adolescent Medicine, School of Clinical Medicine, Li Ka Shing Faculty of Medicine, The University of Hong Kong, Hong Kong SAR, China
5 Department of Paediatrics and Adolescent Medicine, Queen Mary Hospital, Hong Kong SAR, China
6 Department of Paediatrics and Adolescent Medicine, United Christian Hospital, Hong Kong SAR, China
Corresponding author: Dr CW Kong (melizakong@gmail.com)
 Full paper in PDF
Full paper in PDF
Case presentation
A 23-year-old pregnant nulliparous Pakistani
woman underwent first-trimester Down syndrome
screening in July 2019 that deemed her low risk
with normal nuchal translucency. She did not have
any morphology scan. At 34 weeks of gestation her
uterus was considered small-for-dates. Ultrasound
determined that her fetus had intrauterine growth
restriction and multiple abnormalities. There was
severe microcephaly (corresponding to only 24 weeks
of gestation), Dandy-Walker malformation, bilateral
cataracts, micrognathia, abnormal spinal curvature,
and multiple joint contractures with bilateral
clenched hands (Fig 1). On further enquiry, the
patient disclosed that she and her husband were first
cousins. The risk of the fetus having chromosomal
abnormalities or autosomal recessive disease due to
consanguinity was explained to the couple but they
declined amniocentesis. Serial ultrasound scans
showed poor interval growth. Subsequently the
patient had spontaneous onset of labour at 40 weeks
of gestation and delivered a female baby weighing
1.53 kg. The baby was apnoeic and had bradycardia
requiring cardiopulmonary resuscitation for 6
minutes after birth.
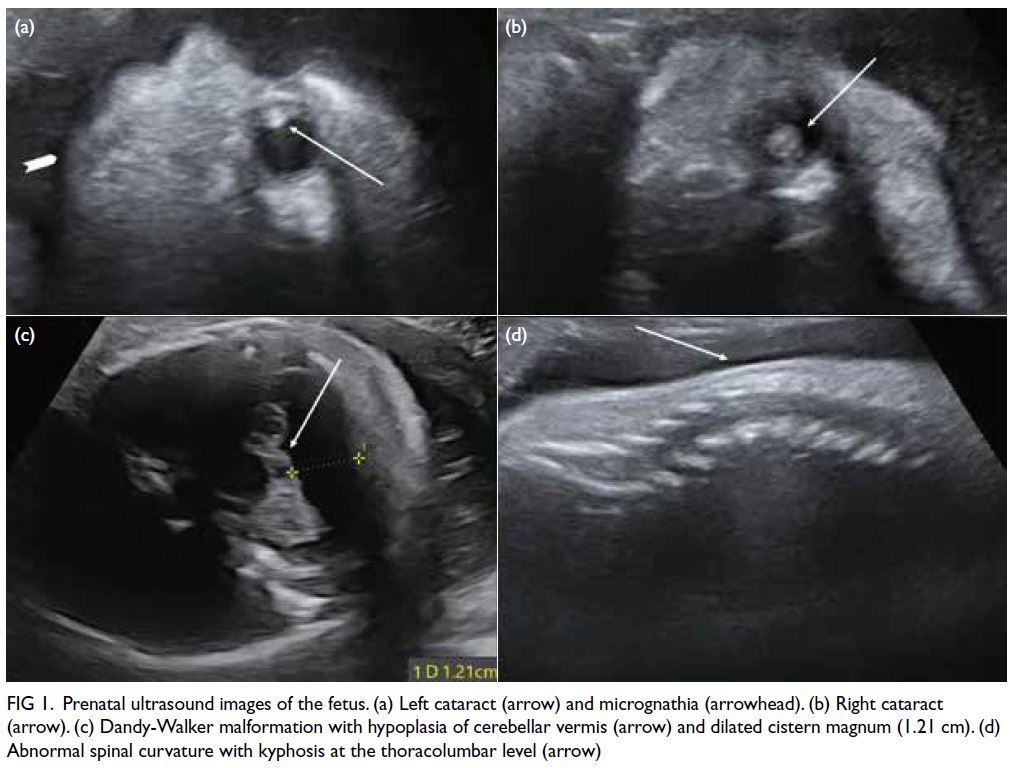
Figure 1. Prenatal ultrasound images of the fetus. (a) Left cataract (arrow) and micrognathia (arrowhead). (b) Right cataract (arrow). (c) Dandy-Walker malformation with hypoplasia of cerebellar vermis (arrow) and dilated cistern magnum (1.21 cm). (d) Abnormal spinal curvature with kyphosis at the thoracolumbar level (arrow)
The baby was confirmed to have multiple
abnormalities similar to the prenatal ultrasound
findings: microcephaly, Dandy-Walker variant,
bilateral dense cataracts and microphthalmia,
persistent arthrogryposis with joint deformities and
contractures, and bilateral lung hypoplasia. Initially
the limbs were oedematous with skin breakages that
subsequently evolved into ichthyosis (Fig 2). Cord
blood was sent for chromosomal microarray after
delivery and showed several regions of >10 Mb long
contiguous stretches of homozygosity consistent with
the known consanguineous relationship of the couple but there were no copy number of changes detected.
Whole exome sequencing (WES) was performed
due to suspected syndromal disease and confirmed
the diagnosis of Neu-Laxova syndrome (NLS) with
homozygous NM_006623.3(PHGDH):c.488G>A
p.(Arg163Gln) missense variant. The parents were
both heterozygous carriers. The child is now >3 years
old (42 months of age at the time of writing). She has
required a tracheostomy and mechanical ventilation
since the age of 6 weeks. She is non-ambulatory
and cannot sit without support. She has hearing
and visual problems and cannot vocalise. She is fed
via a Ryle’s tube and has remained an in-patient in
paediatric intensive care unit since birth.
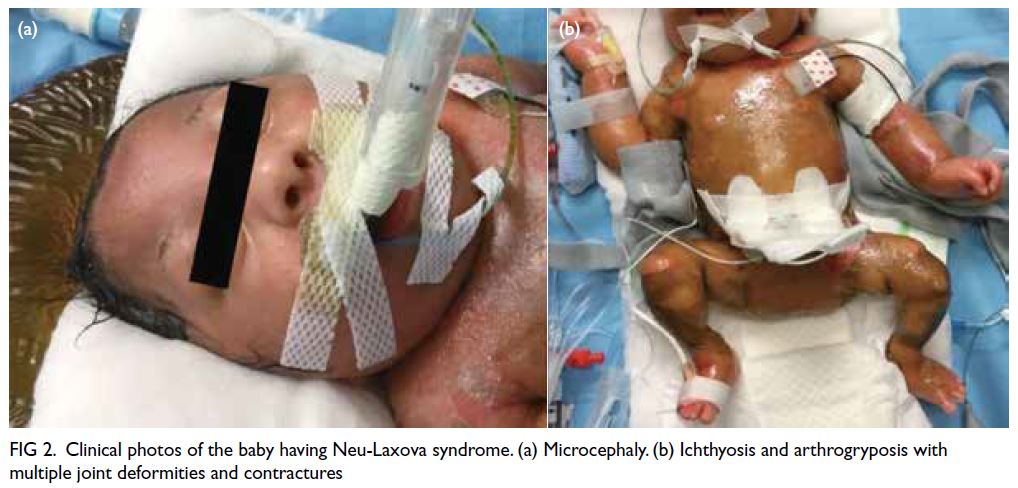
Figure 2. Clinical photos of the baby having Neu-Laxova syndrome. (a) Microcephaly. (b) Ichthyosis and arthrogryposis with multiple joint deformities and contractures
Discussion
Neu-Laxova syndrome is a lethal autosomal
recessive disorder characterised by neuro-oculo-ectodermal
dysplasia with central nervous system
malformations (dominated by microcephaly),
ocular defects (eg, proptosis), craniofacial
dysmorphism (eg, micrognathia, flattened nasal
bridge, and hypertelorism), limb abnormalities
(eg, arthrogryposis), ichthyotic skin changes, and
intrauterine growth restriction. The fetus in our case
had typical features of NLS on prenatal ultrasound
(microcephaly, Dandy-Walker malformation,
ocular defects of congenital cataract, micrognathia,
multiple joint contractures, and intrauterine growth
restriction). This syndrome is caused by mutation
in either one of the three genes involved in the
serine biosynthesis pathway: phosphoglycerate
dehydrogenase (PHGDH), phosphoserine
aminotransferase 1 (PSAT1) or phosphoserine
phosphatase (PSPH) that are essential for synthesis
of brain lipids. Up to 2022, 88 cases of NLS had been
reported with 45% related to consanguinity.1 The early cases reported were diagnosed clinically and
by histopathological examination and following the
emergence of molecular genetic diagnosis in 2014 by WES.1
Chromosomal microarray can detect only copy number changes (gains or losses), not gene
mutations. This case illustrates the usefulness
of molecular diagnosis by WES for a baby with
multiple congenital abnormalities. The diagnosis
can be quickly established and the prognosis of the baby can be explained to the parents with
appropriate supportive counselling. Molecular
diagnosis is also helpful to verify parental carrier
status so that the risk for future pregnancies can be
predicted. In this case, with both parents a carrier,
the risk of recurrence in any future pregnancy is
25%. Prenatal diagnosis by chorionic villus sampling
or amniocentesis should be offered, and the option
of termination of pregnancy should be discussed if
the fetus is affected. Alternatively, pre-implantation
genetic testing can be offered to select unaffected
embryos and avoid a recurrently affected pregnancy.
Since April 2021, a publicly funded prenatal genomic
sequencing programme for antenatally diagnosed
fetal structural anomalies has been available in the
Hospital Authority. Pregnant women are considered
eligible if the chromosomal microarray results are
normal and if fetal abnormalities are considered by
an expert panel to have high possibility of genetic
aetiology.2 Although publicly funded postnatal
genomic sequencing is available for newborn babies
and children, the test is usually performed only in
highly selected cases and the turnaround time is often
substantial. The WES in our case was performed by
The University of Hong Kong in a research setting.
There will be a need to enhance the provision of such
services to meet the expanding clinical demands.
A local study found that consanguineous
couples have an increased risk of autosomal
recessive diseases in their offspring with an odds
ratio of 8.7.3 A mid-trimester morphology scan
should be performed to screen for fetal structural
anomalies, and expanded carrier screening is advised
for consanguineous couples prior to conception.
Different pregnancy options can then be discussed,
including pre-implantation genetic testing if both
parents are a carrier of the same autosomal recessive
disease. The expanded carrier screening panel that
is available in Hong Kong includes the PHGDH gene
responsible for NLS in our patient. If such screening
had been performed before conception, the carrier
status of the couple would have been identified. The
observed positive carrier frequency of autosomal
recessive diseases in the Chinese population has
been reported as high as 58.7% overall (47.6%
after excluding thalassaemias) in a local cohort.4 If
resources allow, expanded carrier screening can be
offered to all couples who wish to conceive, even in
the absence of known consanguinity.
Neu-Laxova syndrome is a lethal disorder with
most affected babies stillborn or dying soon after
birth. The longest survival previously reported is 10
months of age.5 A case report in 2013 described a
collodion baby who survived to 8 years of age with
facial dysmorphism, limb anomalies, pachygyria and genital hypoplasia, but without the most common
features of microcephaly or intrauterine growth
restriction, who was diagnosed clinically to have a
mild form of NLS without molecular diagnosis.5 Our
patient has the longest survival among the confirmed
NLS cases reported in the literature.
In conclusion, WES can help establish a quick
and accurate diagnosis of NLS in a baby with multiple
structural abnormalities. Expanded carrier screening
is advised for at-risk couples before conception to
verify their carrier status and enable counselling
about future pregnancy risks and options.
Author contributions
Concept or design: CW Kong.
Acquisition of data: YY Li.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: CW Kong.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: YY Li.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: CW Kong.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study was supported by the Health and Medical Research
Fund of Health Bureau of the Hong Kong SAR Government
and The Society for the Relief of Disabled Children (Ref No.:
06172806). The funders had no role in study design, data
collection/analysis/interpretation or manuscript preparation.
Ethics approval
The patient was treated in accordance with the Declaration of
Helsinki. Written consent was obtained for publication of this
article and accompanying images.
References
1. Serrano Olave A, López AP, Cruz MM, Rodríguez SM, Narbona Arias I, López JS. Prenatal diagnosis of Neu-Laxova syndrome. Diagnostics (Basel) 2022;12:1535. Crossref
2. So PL, Hui AS, Ma TW, et al. Implementation of public
funded genome sequencing in evaluation of fetal structural
anomalies. Genes (Basel) 2022;13:2088. Crossref
3. Siong KH, Au Yeung SK, Leung TY. Parental consanguinity
in Hong Kong. Hong Kong Med J 2019;25:192-200. Crossref
4. Chan OY, Leung TY, Cao Y, et al. Expanded carrier
screening using next-generation sequencing of 123 Hong
Kong Chinese families: a pilot study. Hong Kong Med J
2021;27:177-83. Crossref
5. Ozcan D, Derbent M, Seçkin D, et al. A collodion baby
with facial dysmorphism, limb anomalies, pachygyria and
genital hypoplasia: a mild form of Neu-Laxova syndrome
or a new entity? Ann Dermatol 2013;25:483-8. Crossref