Hong Kong Med J 2024 Feb;30(1):63–5 | Epub 8 Feb 2024
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Multisystemic smooth muscle dysfunction syndrome: the first local case report
CH Ng, MB, ChB, MRCPCH; Grace PK Chiang, MSc, FHKAM (Paediatrics); KW Tsui, MRCP, FHKAM (Paediatrics)
Department of Paediatrics and Adolescent Medicine, Alice Ho Miu Ling Nethersole Hospital, Hong Kong SAR, China
Corresponding author: Dr CH Ng (nch234@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Case presentation
In February 2020, a 7-year-old girl was brought
to our institution. She had been experiencing
dysphasia, dysphagia, and weakness in all four limbs
for the past 5 days. She had no recent trauma, febrile
illness or seizure. She had normal systolic blood
pressure but diastolic pressure was persistently low
for her age at 35 to 40 mm Hg. She had a Glasgow
Coma Scale score of 11 (E4V1M6) with expressive
aphasia. Pupils were equal but dilated at 5 mm
and cranial nerves were grossly intact. The muscle
strength of right lower limb power was the weakest,
with a Medical Research Council (MRC) grade of
3, while others were at grade 4. She had brisk jerks
over the lower limbs. Her medical history included
premature birth at 34 weeks of gestation and surgical
repair of a patent ductus arteriosus at 1 month of age.
She also had recurrent urinary tract infections with
megacystic bladder, managed since early infancy
with intermittent bladder catheterisation. Magnetic
resonance imaging of the brain was performed at the
age of 5 years due to chronic headache and showed a
large area of right frontal encephalomalacia that may
have been due to a previous unknown insult.
Blood tests including complete blood count,
electrolytes, inflammatory markers, lactate, ammonia, dried blood spot test, homocysteine, and
plasma amino acids were unremarkable. Clotting
profile, protein C, protein S and antithrombin III, and
antinuclear antibodies levels were normal. Magnetic
resonance imaging of the brain revealed acute
ischaemic stroke with diffusion restriction in the left
superior frontal, precentral, cingulate cortices, and
corona radiata. Magnetic resonance angiography
showed the anterior cerebral artery and middle
cerebral artery were replaced by pruned cerebral
arteries with straightened appearance and multifocal
stenosis (Fig 1). Transthoracic echocardiogram
showed a dilated aortic root of 23 mm with mild
aortic regurgitation. Magnetic resonance aortogram
showed comparable measurement with no aortic
aneurysm or dissection (Fig 2).
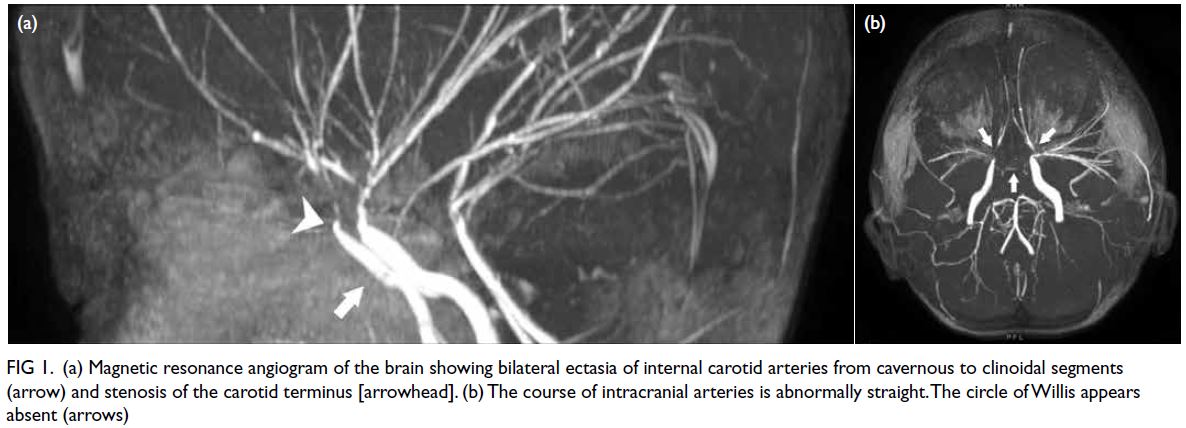
Figure 1. (a) Magnetic resonance angiogram of the brain showing bilateral ectasia of internal carotid arteries from cavernous to clinoidal segments (arrow) and stenosis of the carotid terminus [arrowhead]. (b) The course of intracranial arteries is abnormally straight. The circle of Willis appears absent (arrows)
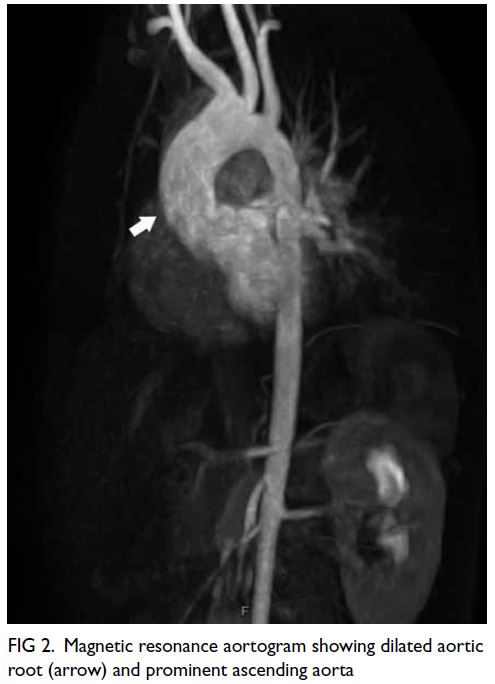
Figure 2. Magnetic resonance aortogram showing dilated aortic root (arrow) and prominent ascending aorta
To account for all clinical manifestations of
the patient, a rare condition, multisystemic smooth
muscle dysfunction syndrome (MSMDS), was
suspected. Genetic analysis confirmed the diagnosis
with a de novo heterozygous mutation of ACTA2
c.536G>A (Arg179His). No similar mutation was
identified in her parents or younger sister.
Our patient was managed conservatively and
prescribed oral aspirin. There have been five further
episodes of ischaemic stroke or transient ischaemic attack to date. Developmental assessments revealed
mild-grade intellectual disability with regression
of gross and fine motor skills. During the latest
follow-up, she could walk unaided and manage oral
feeding. Physical examination showed grossly full
proximal power. The right-hand flexors were weak
(Medical Research Council grading of 4/5) and
right lower limb was spastic. The patient is being
followed by a multidisciplinary team comprised of a
neurologist, cardiothoracic surgeon, neurosurgeon,
and ophthalmologist.
Discussion
To the best of our best knowledge, this is the first
identified case of MSMDS in Hong Kong. The
ACTA2 gene encodes the thin filaments alpha-actin
of the contractile element of smooth muscle. In
2010, Milewicz et al1 reported the first case series
of patients with de novo missense mutation in the
ACTA2 gene. Arg179His is the most severe form of
this vascular disease spectrum. The three cardinal
clinical presentations are congenital mydriasis,
patent ductus arteriosus/aortopulmonary window,
and white matter lesions with cerebrovascular
disease.
Less common mutation loci, including Arg179Cys, Arg179Leu and Arg179Ser, have been reported to cause MSMDS.2
Neurological aspect
Recurrent ischaemic stroke is the major debilitating
manifestation in MSMDS patients. Up to 95% of
these patients have periventricular white matter
hyperintensities evident on magnetic resonance
imaging, signifying ischaemic insult in these
watershed areas of blood supply. The vasculopathies
were once considered a variant of moyamoya
disease. Nonetheless MSMDS has some unique
features, namely dilatation from the cavernous
to the clinoid segments of the internal carotid
arteries, stenosis and occlusive disease of the distal
intracranial circulation, and an abnormally straight
course of intracranial arteries. Stenosis of major
cerebral arteries arises as a result of significant
intimal thickening and markedly increased collagen
and smooth muscle cells in the medial layers of these
vessels.3
Neurosurgical management similar to that
applied in moyamoya disease has been extrapolated
to patients with MSMDS and includes direct
superior temporal artery to anterior cerebral artery
revascularisation and indirect revascularisation
such as encephaloduroarteriosynangiosis.
Nonetheless patients with various ACTA2 mutations
respond less well to these operations. Almost
half of affected patients who have undergone
neurosurgical revascularisation continue to have
major stroke episodes.4 After a case conference with
neurosurgeons, anaesthesiologists, neurologists
and the parents of our patient, we concluded
that as surgery carried an exceedingly high risk
with doubtful benefit, revascularisation was not
considered.
Cardiovascular aspect
All reported cases of MSMDS have had a patent
ductus arteriosus or aortopulmonary window with
most requiring surgical ligation. Patent ductus
arteriosus with diameter up to 20 mm in a neonate
with MSMDS has also been reported.
Regalado et al2 reported that among the
various ACTA2 mutations, Arg179His carries the
highest risk of serious aortic events. By the age
of 25 years, the cumulative risk of either elective
aortic aneurysm repair or dissection is 100%.5 The
group also suggested that elective surgical repair be
considered when the aortic diameter reaches 4.5 cm.5
The benefit of drug therapy such as beta-blockers or
angiotensin receptor blockers is unknown. It was not
appropriate to prescribe either drug for our patient
since further lowering of blood pressure would
aggravate cerebral hypoperfusion leading to more
stroke episodes.3
Apart from aortopathy, around half of MSMDS
patients have peripheral arterial dilation involving
the proximal internal carotid artery, common carotid
brachiocephalic, subclavian, axillary and external/internal iliac arteries. This might also explain the low diastolic pressure.
Other systems
All reported cases of MSMDS have had pupillary
abnormalities, mostly fixed or nonreactive pupils.
Iris hypoplasia or aniridia has also been reported.
Pulmonary complications, though not yet evident
in our case, are also common. Pulmonary arterial
hypertension may present in early infancy and
necessitate mechanical ventilation, vasodilator drugs
or even lung transplantation. Asthma and chronic
lung disease have also been reported.
About half of MSMDS patients have hypotonic
bladders. Recurrent urinary tract infections and
hydronephrosis, as in our case, have also been
frequently identified. One-third of patients have
gastrointestinal problems such as gut malrotation,
gastroesophageal reflux disease or constipation.2
In conclusion, MSMDS can affect multiple
systems with life-threatening complications. Patients
with congenital mydriasis, patent ductus arteriosus
and recurrent stroke may raise a clinician’s suspicion
of this rare condition and prompt early genetic
investigations.
Author contributions
Concept or design: CH Ng.
Acquisition of data: CH Ng.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: CH Ng.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: All authors.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of Helsinki and provided informed consent for all treatments and procedures, and consent for publication.
References
1. Milewicz DM, Østergaard JR, Ala-Kokko LM, et al.
De novo ACTA2 mutation causes a novel syndrome of
multisystemic smooth muscle dysfunction. Am J Med
Genet A 2010;152A:2437-43. Crossref
2. Regalado ES, Mellor-Crummey L, De Backer J, et al.
Clinical history and management recommendations of
the smooth muscle dysfunction syndrome due to ACTA2
arginine 179 alterations. Genet Med 2018;20:1206-15. Crossref
3. Georgescu MM, Pinho M da C, Richardson TE, et al. The
defining pathology of the new clinical and histopathologic
entity ACTA2-related cerebrovascular disease. Acta
Neuropathol Commun 2015;3:81. Crossref
4. Cuoco JA, Busch CM, Klein BJ, et al. ACTA2 cerebral arteriopathy: not just a puff of smoke. Cerebrovasc Dis 2018;46:159-69. Crossref
5. Regalado ES, Guo DC, Prakash S, et al. Aortic disease presentation and outcome associated with ACTA2 mutations. Circ Cardiovasc Genet 2015;8:457-64. Crossref