Hong Kong Med J 2023 Aug;29(4):351–4 | Epub 12 Jul 2023
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Novel compound heterozygous mutation of the diacylglycerol kinase epsilon gene and membranoproliferative glomerulonephritis: a case report
Sharon HY Lau, MB, ChB, MRCPCH1; Eugene YH Chan, FHKAM (Paediatrics)2; Liz YP Yuen, FHKCPath, FHKAM (Pathology)3; WF Ng, FHKAM (Pathology)3; Alison LT Ma, FHKAM (Paediatrics)2
1 Department of Paediatrics, Prince of Wales Hospital, Hong Kong SAR, China
2 Paediatric Nephrology Centre, Department of Paediatrics, Hong Kong Children’s Hospital, Hong Kong SAR, China
3 Department of Pathology, Hong Kong Children’s Hospital, Hong Kong SAR, China
Corresponding author: Dr Eugene YH Chan (eugene.chan@ha.org.hk)
 Full paper in PDF
Full paper in PDF
Case report
A 10-year-old Chinese boy first presented in June
2015 at the age of 4 years with steroid-resistant
nephrotic syndrome. He had been taking full-dose
prednisolone at 60 mg/m2/day for 4 weeks. Renal
biopsy confirmed immune complex–mediated
membranoproliferative glomerulonephritis (MPGN)
with predominant immunoglobulin M (IgM)
and scanty IgG or C3 deposits. Cyclosporin A, a
calcineurin inhibitor (CNI), was introduced and
successfully brought the disease into partial remission.
His urine protein–to-creatinine ratio (UPCR)
reduced from 13.9 mg/mg to 0.95 mg/mg after 2
months. He remained in partial remission for 4 years
with cyclosporin A and low-dose prednisolone. He
showed a urinary relapse following discontinuation
of cyclosporin A in 2020 with a rebound in UPCR
from 0.64 mg/mg to 1.57 mg/mg (Table).
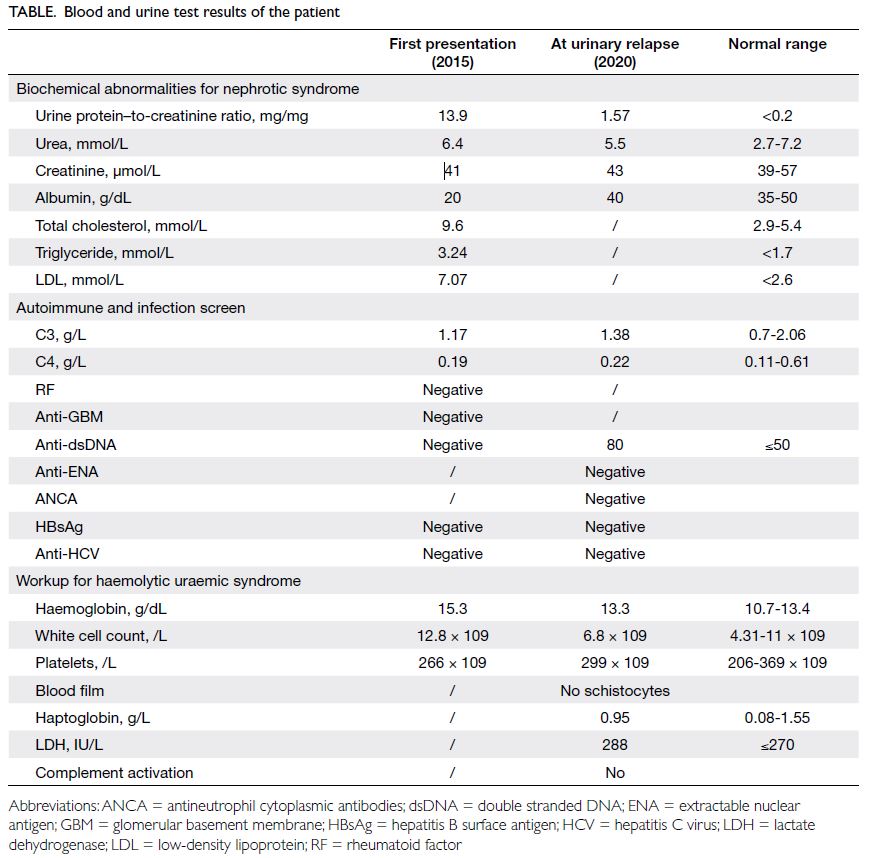
Table. Blood and urine test results of the patient
Renal biopsy was repeated and revealed mild-to-moderate global mesangial cell proliferation
and a few clusters of arterioles showing hyaline
arteriolosclerosis with a peripheral and segmental
distribution (Fig). Of note, there was evidence of
mesangiolysis with loss of argyrophilic basement
membrane material in a segmental pattern. The
direct immunofluorescence portion showed
finely granular deposits of IgA (2+), IgG (3+), IgM
(2+), C1q (3+), and C3 (+) in a diffuse global and
capillary distribution. Electron microscopy showed
focal fusion of podocyte foot processes, splitting
of glomerular basement membrane in association
with mesangial cell interposition and scattered
subendothelial and intramembranous dense
electron deposits in keeping with immune complex.
The features were indicative of immune complex–mediated MPGN with evidence of CNI toxicity. The
presence of segmental mesangiolysis was suggestive
of previous endothelium injury, possibly an episode
of glomerular thrombotic microangiopathy (TMA).
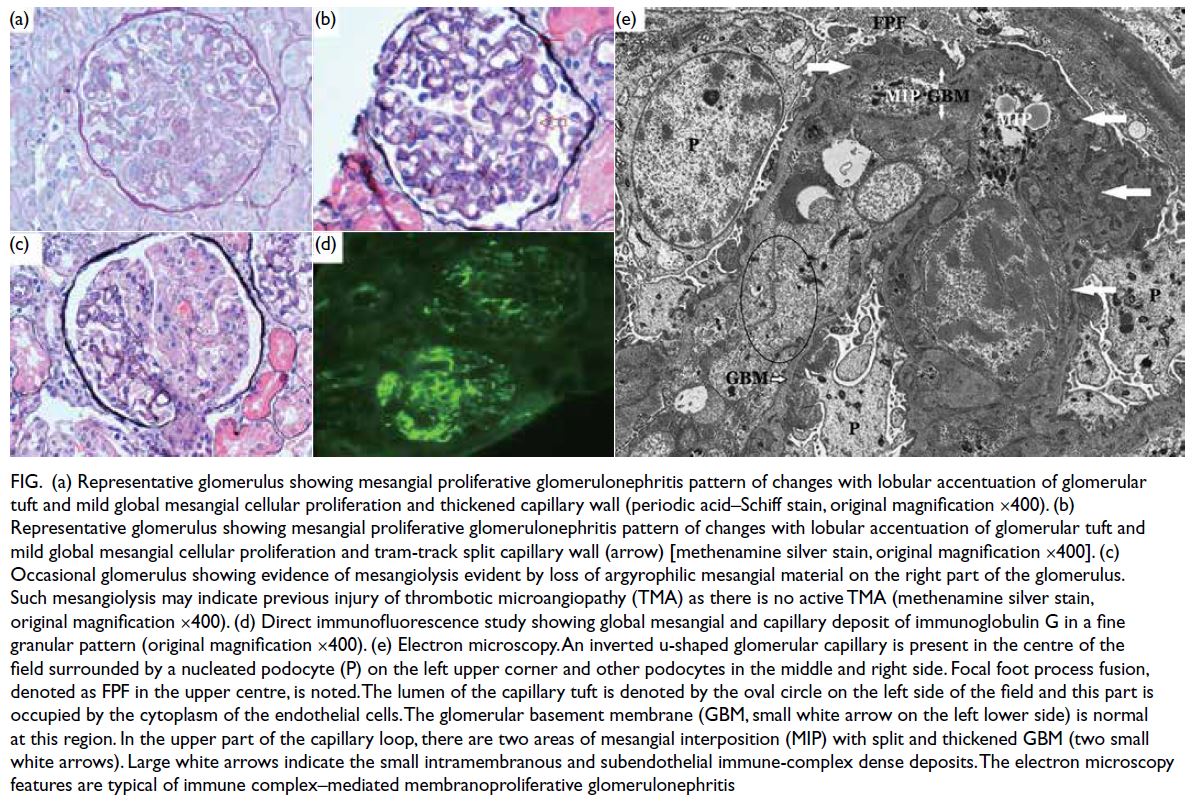
Figure. (a) Representative glomerulus showing mesangial proliferative glomerulonephritis pattern of changes with lobular accentuation of glomerular tuft and mild global mesangial cellular proliferation and thickened capillary wall (periodic acid–Schiff stain, original magnification ×400). (b) Representative glomerulus showing mesangial proliferative glomerulonephritis pattern of changes with lobular accentuation of glomerular tuft and mild global mesangial cellular proliferation and tram-track split capillary wall (arrow) [methenamine silver stain, original magnification ×400]. (c) Occasional glomerulus showing evidence of mesangiolysis evident by loss of argyrophilic mesangial material on the right part of the glomerulus. Such mesangiolysis may indicate previous injury of thrombotic microangiopathy (TMA) as there is no active TMA (methenamine silver stain, original magnification ×400). (d) Direct immunofluorescence study showing global mesangial and capillary deposit of immunoglobulin G in a fine granular pattern (original magnification ×400). (e) Electron microscopy. An inverted u-shaped glomerular capillary is present in the centre of the field surrounded by a nucleated podocyte (P) on the left upper corner and other podocytes in the middle and right side. Focal foot process fusion, denoted as FPF in the upper centre, is noted. The lumen of the capillary tuft is denoted by the oval circle on the left side of the field and this part is occupied by the cytoplasm of the endothelial cells. The glomerular basement membrane (GBM, small white arrow on the left lower side) is normal at this region. In the upper part of the capillary loop, there are two areas of mesangial interposition (MIP) with splitted and thickened GBM (two small white arrows). Large white arrows indicate the small intramembranous and subendothelial immune-complex dense deposits. The electron microscopy features are typical of immune complex–mediated membranoproliferative glomerulonephritis
Next-generation sequencing was performed
and detected two mutations in the diacylglycerol kinase epsilon (DGKE) gene, including a
c.1068_1071del p.(Asn356Lysfs*6) frameshift
mutation and a c.1282_1284+18del deletion. Sanger
sequencing of the mother detected heterozygous
DGKE gene c.1068_1071del p.(Asn356Lysfs*6).
The father was unavailable for genetic analysis. In
view of these results, the two DGKE variants were
likely to act in trans in the patient. According to the
American College of Medical Genetics/Association
for Molecular Pathology classification, these two
variants are considered pathogenic. Additional
whole-exome sequencing revealed no significant
variants in genes related to monogenic lupus.
Throughout the course of the disease, his blood
tests showed no features of haemolytic uraemic
syndrome (HUS). Apart from a mildly elevated lactate
dehydrogenase, his complete blood count, blood
smear and haptoglobin were normal. Complement
function testing showed no evidence of complement
activation. He was commenced on an angiotensin-converting
enzyme inhibitor, prednisolone, and
mycophenolate mofetil. He responded promptly
with marked improvement in proteinuria: UPCR
decreased to 0.36 mg/mg within 1 month. At last
follow-up, his UPCR was 0.24 mg/mg with normal
kidney function (estimated glomerular filtration
rate = 116 mL/min/1.73 m2).
Discussion
We report the first Chinese patient with DGKE
nephropathy who presented as nephrotic syndrome
and immune complex–mediated MPGN. Importantly,
the patient responded well to immunosuppressive
agents including CNI and mycophenolate mofetil. To
the best of our knowledge, the pathogenic variants
in this case are the first reported in DGKE-related
nephrotic syndrome.
Diacylglycerol kinase epsilon gene is important
in the regulation of thrombotic status and is present
in podocytes, platelets, and endothelium. A loss-of-function in the DGKE gene is associated with a
prothrombotic state, leading to episodes of HUS
that are complement-independent.1 Interestingly,
a subset of patients develop MPGN with nephrotic
syndrome.2 Along with the new classification, it is
believed that chronic microangiopathy often results
in a form of MPGN that is neither immune complex–mediated nor complement-mediated.3 The exact
mechanism of DGKE mutations leading to MPGN
remains unknown.
Azukaitis et al4 reported 44 cases of DGKE
nephropathies, including 33 cases with HUS,
nine cases with MPGN/nephrotic syndrome,
and two rare cases with a mixed HUS/MPGN presentation. Among the nine reported cases of DGKE-MPGN, three other pathogenic variants
were identified. Mutations of p.Gln43*, p.IVS5-2a
and p.Thr204Glnfs*6 were found in four, three and
two of the patients, respectively. Interestingly, the
first case of DGKE nephropathy confirmed in our
territory-wide next-generation sequencing cohort
had nephrotic syndrome and MPGN.
The pathology of renal biopsy in this child is
particularly interesting in terms of the presence of
TMA features and an immune complex–mediated
MPGN picture. The presence of mesangiolysis
signifies endothelial injury such as TMA. These
lesions might be caused by the underlying disease
process related to DGKE mutations. Of note, about
half of the patients with DGKE-HUS recovered
spontaneously from the initial TMA episode without any HUS-specific treatment.4 A potential
explanation of this observation in our patient is that
an episode of subclinical TMA had resolved on its
own. Another interesting finding was the positive
immunofluorescence on renal biopsy, not commonly
seen in TMA-related MPGN. The immune-complex
deposition in our patient could not be explained by
either autoimmune disease or hepatitis. Although he
had an isolated elevation of anti–double stranded
DNA that occurred only on disease relapse, a
confirmed diagnosis of systemic lupus could not
be excluded. Whole-exome sequencing revealed
no clinically significant variants in the monogenic
lupus genes. Whether or not the finding of immune
complex deposition at a histological level was an
event secondary to the genetic mutation remains
unknown. Clinicians need to be mindful of such
a discrepant genetic finding and the clinical
phenotype with close follow-up. There have been
reported cases of patients with TMA who developed
immune complex–mediated MPGN, one of whom
also carried a DGKE mutation.4 5 The pathogenic and
prognostic differences of this immune complex–mediated subgroup require further elucidation.
Regarding prognosis and treatment, most
patients with DGKE-MPGN show some response to
immunosuppressants. Nonetheless Azukaitis et al4
reported a high prevalence of chronic proteinuria
in DGKE-MPGN patients. Although statistically
insignificant, a trend towards a lower 10-year renal
survival was also observed (50% in DGKE-MPGN
vs 89% in DGKE-HUS).4 After initiation of CNI
or mycophenolate mofetil, our patient showed a
promising response and attained remission. At
5-year follow-up, he remained in remission with
normal kidney function.
This is the first Chinese patient with DGKE
nephropathy presenting as nephrotic syndrome
with an MPGN picture. Of interest, our patient
responded promptly to immunosuppressants,
suggesting its potential role in this disease entity.
This case highlights the importance of genetic
testing in children with an atypical course of
nephrotic syndrome. Further international
collaborative studies are warranted to understand the
pathogenesis and optimal management in this patient population.
Author contributions
Concept or design: SHY Lau, EYH Chan.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: SHY Lau, EYH Chan.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: SHY Lau, EYH Chan.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Acknowledgement
We thank the Department of Pathology, Pamela Youde
Nethersole Eastern Hospital for their permission to review
the patient’s first renal biopsy.
Declaration
This case was accepted as poster presentation in the Joint Annual Scientific Meeting 2021 of The Hong Kong Paediatric
Society, Hong Kong College of Paediatricians, Hong Kong
Paediatric Nurses Association and Hong Kong College of
Paediatric Nursing (hybrid, 30 October 2021).
Funding/support
This study received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of Helsinki, with informed consent provided by the patient’s
mother for treatment, procedures, and publication.
References
1. Lemaire M, Frémeaux-Bacchi V, Schaefer F, et al. Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 2013;45:531-6. Crossref
2. Ozaltin F, Li B, Rauhauser A, et al. DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J Am Soc Nephrol 2013;24:377-84. Crossref
3. Masani N, Jhaveri KD, Fishbane S. Update on
membranoproliferative GN. Clin J Am Soc Nephrol 2014;9:600-8. Crossref
4. Azukaitis K, Simkova E, Majid MA, et al. The phenotypic spectrum of nephropathies associated with mutations in
diacylglycerol kinase ε. J Am Soc Nephrol 2017;28:3066-75. Crossref
5. Ankawi GA, Clark WF. Atypical haemolytic uremic syndrome (aHUS) and membranoproliferative glomerulonephritis (MPGN), different diseases or a spectrum of complement-mediated glomerular diseases? BMJ Case Rep 2017;2017:bcr2017220974. Crossref