© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
COMMENTARY
Lessons learnt from a genetic disease registry in
Hong Kong
Stephen TS Lam, MD, FHKAM (Paediatrics)1 CH To, PhD2; KW Leung, FCOphth HK, FRCOphth2; SP Yip, PhD3; Ivan FM Lo, FHKCPaed, FHKAM (Paediatrics)4; KP Tsang5
1 Department of Clinical Genetics Service, Hong Kong Sanatorium & Hospital, Hong Kong
2 School of Optometry, The Hong Kong Polytechnic University, Hong Kong
3 Department of Health Technology and Informatics, The Hong Kong Polytechnic University, Hong Kong
4 Clinical Genetic Service, Department of Health, Hong Kong
5 Retina Hong Kong, Hong Kong
Corresponding author: Prof Stephen TS Lam (taksumlam@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Purpose of a genetic disease registry
A registry for a specific clinical condition
serves multiple purposes. Firstly, it provides a
systemic index of patients and families with the
condition. Secondly, it helps in the classification
and management of patients with the condition.
Thirdly, it is a good resource for research into the
epidemiology, phenotype and genotype variations,
disease mechanisms, and treatment modalities of
the condition. Fourthly, from the above functions,
it works as a tool for advocacy for the welfare of
patients with the condition. To serve these purposes
for patients with retinitis pigmentosa (RP) in Hong
Kong, a registry was started, and the following is a
summary of the issues encountered and lessons learnt.
Retinitis pigmentosa registry in
Hong Kong
A rare genetic eye disorder, RP causes progressive
visual loss due to degeneration of cells in the
retina, estimated to affect 1 in 4000 people in the
United States and worldwide.1 There are, however,
no reported prevalence data in Hong Kong. The
diagnosis of RP is based on the clinical features
which include symptoms of night vision impairment
and tunnel vision, and the findings of typical retinal
appearance of attenuated blood vessels, so-called
bone spicule pigmentation or its variant appearance
and an atrophic optic disc. These are supported
by an abnormal constricted visual field and a nonrecordable
or abnormal electroretinography with
diminished a and b waves with increased implicit
time. The RP is also highly genetically heterogeneous,
with more than 80 genes implicated and their mode
of inheritance varies. This means one patient’s
RP may be a different genetic disorder from that
of another RP patient, with different inheritance,
natural course, and prognosis. The rarity of the
disease and the associated genetic heterogeneity are major obstacles to facilitating research and
enhancing medical care to these patients and their
families. The Hong Kong Retinitis Pigmentosa
Society was started in 1995 as a patient group, and
subsequently renamed as Retina Hong Kong. Under
the auspices of Retina Hong Kong, a Scientific
and Medical Advisory Committee (SMAC) was
established. In 1997, the SMAC secured a grant from
the Healthcare and Promotion Fund of the Hong
Kong Government to start organising the Hong
Kong Patients Registry of Retinal Degenerations. It
was the original intention for this registry to cover
most retinal degenerative conditions. However,
because of resource constraints, it was decided that
the scope should be limited to RP as the first group of
members to be covered. Hence, the registry was also
abbreviated as the Hong Kong Retinitis Pigmentosa
Registry. It turned out to be the first registry of its
kind among Chinese populations in the whole world,
and to the best of our knowledge, there is no other
Chinese Registry presently.
The objectives of the Retinitis Pigmentosa
Registry are three-fold, including:
To provide detailed ophthalmic and genetic
examinations for inherited retinal degenerative
diseases patients so as to ascertain the symptoms
and the genetic pattern of the patients.
To collect relevant data and build up a database
for the benefit of future scientific, medical, and
sociological researches.
To provide lifelong follow-up services for patients
who participate in the project and to notify and
refer them for appropriate treatment as soon as it
is available.
The holder of the Registry is the Retina Hong
Kong, whose members were offered the option
of voluntary participation in the Registry. Patient
members were invited to be seen by the specialists in
the SMAC in a sequence as outlined in the flowchart
in the Figure for clinical examinations, diagnostic
testing, and counselling.
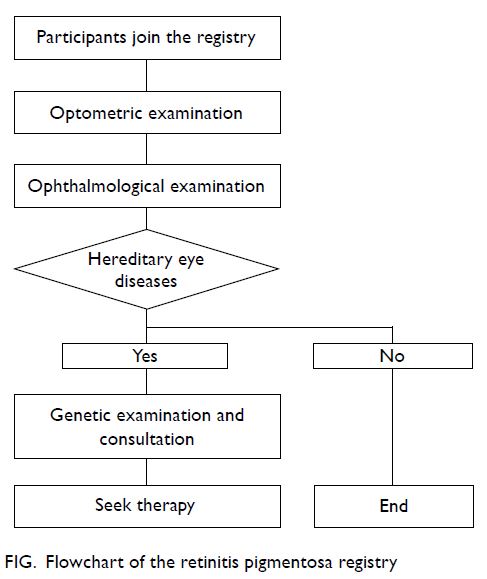
Figure. Flowchart of the retinitis pigmentosa registry
Right at the start of their participation,
members were advised of the detailed flow of the
whole process. More importantly, they were advised
of their rights in this participation before signing for
consent. Their rights included:
To obtain detailed ophthalmic examination and
eye care apart from advice on genetics.
To know well their condition and progress
through different clinical examinations.
To be informed of all findings that might help in
their treatment plan.
To make use of relevant information to formulate
their future plan on career and family.
The Registry is maintained strictly in accordance
with the Personal Data (Privacy) Ordinance.
Participants may obtain all information relating
to their own case, which is stored in the database of the
Registry.
To ensure that participants are well informed
of the flow of the registry and their rights and
obligations, pamphlets were designed and made
available as part of the informed consent process.
The first clinical assessment for the
participants was an optometric evaluation at The
Hong Kong Polytechnic University (HKPU), where
refraction studies, ocular health assessment, visual
field test, electroretinogram studies, and fundus
photography were performed. This was followed by
an ophthalmologic examination with dilated fundus
evaluation. As shown in the Figure, eye diseases are
defined upon clinical criteria as to whether they
fall into the categories of known hereditary nature. The third clinical assessment was genetic evaluation
and counselling together with sample collection
(blood sample with DNA extraction) performed at
the Clinical Genetic Service of the Department of
Health. Further genetic testing was carried out at
the molecular laboratory of HKPU for mutational
analysis. Periodic clinical and molecular conferences
were conducted among members of the SMAC to
confirm the exact clinical diagnosis, genetic defects,
and inheritance patterns for individual participants
to guide further genetic counselling, whence other
family members were also invited to participate on a
voluntary basis.
Periodic assessment of the Registry was
conducted by the SMAC and reported to the Retina
Hong Kong. The first of these reports covered the
period 1999 to 2005, during which a total of 94
families were documented. After detailed evaluation
was conducted conjointly by the specialists in the
SMAC, the diagnosis of RP was documented in
72 of these families with inheritance pattern
classified as simplex (44, 61.1%), autosomal dominant
(12, 16.7%), autosomal recessive (12, 16.7%) and
X-linked recessive (4, 5.6%), which showed
comparable results to those published by other
centres2 3; and the remaining 22 families of alternative
diagnosis with various genetic backgrounds. It
was noted that the phenotype of patients with
non-RP retinal degenerations may overlap with
that of RP, leading to erroneous clinical diagnosis,
as exemplified in 30% of this cohort. By combining
clinical data and genetic analysis, a more definitive
diagnosis could be achieved.
Cases of RP and other retinal degenerative
diseases were categorised and published.4 5 In these
early days of molecular analysis, genetic studies were
carried out on putative genes individually, hence on
a gene by gene basis, instead of on a multiplex basis.
As expected, the positive yield of such a laborious
approach was necessarily low.
Funding issues
It was most fortunate that the Healthcare Promotion
Fund has sponsored this registry for nearly 5 years
(1997-2002). With the cessation of this sponsorship,
the Registry could not be sustained, and was
suspended in 2002. It was resumed in April 2003
after further sponsorship was obtained from Retina
Hong Kong, with additional financial input from the
individual institutes to which the members of SMAC
were affiliated.
Further progress of the registry
With the advent of the massively parallel sequencing
(also known as next generation sequencing)
technology, it became possible for mutational
analysis to be performed on a multiplex instead of single gene basis. The previous cohort examined
until 2002 was expanded with new entries to a total
of 116 families by 2012. Their samples were subjected
to next generating sequencing performed on MiSeq
(Illumina) system, initially at HKPU, and, as an
ongoing process, also in Clinical Genetic Service
of the Department of Health. Additional mutations
were identified and put to good use in genetic
counselling of the affected individuals and families.
Lessons learnt
The above narration has highlighted some issues
pertaining to the success or failure of this kind
of registries. The Retinitis Pigmentosa Registry
is notably the first of its kind among all Chinese
populations, and hence has set an exemplary
function for others. The unique feature of the
Retinitis Pigmentosa Registry is that it is initiated
and led by a patient organisation, which works for
the welfare of its members. As such, the interest
of the patients and their families were considered
the primary objective of the registry. All necessary
steps were taken to upkeep the ethical principles
for patient confidentiality and informed consent
as exercised in the design and implementation of
this registry. In addition, a central core of highly
motivated individuals and institutes from the
clinical and scientific community is needed to
provide the academic and service input to make it
viable. From this combined clinical and scientific
approach, an accurate and reliable diagnosis
could be provided for genetic counselling of many
subjects. The data attained from the studies had also
provided information for the spectrum of retinal
degenerations seen in Chinese populations.
Yet, this registry has also uncovered a
major problem with setting up genetic registries:
financial instability. The experience shown here
is that a strategy is needed to ensure the financial
sustainability of the project. To achieve the objectives
of the provision of ongoing care for the participants,
a secure source of funding is needed on a long-term basis. It is advisable to explore different sources of
financial commitment both in the public and private
sectors.
Author contributions
All authors contributed to the concept or design of the study, acquisition of data, analysis or
interpretation of data, drafting of the manuscript, and critical
revision of the manuscript for important intellectual content.
All authors had full access to the data, contributed to the
study, approved the final version for publication, and take
responsibility for its accuracy and integrity.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Funding/support
The funders mentioned in the article had no role in study design, data collection/analysis/interpretation or manuscript
preparation.
References
1. National Eye Institute. Retinitis pigmentosa. Available
from: https://www.nei.nih.gov/learn-about-eye-health/eye-conditions-and-diseases/retinitis-pigmentosa. Accessed 1 Jan 2021.
2. Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS,
Parmeggiani F. Retinitis pigmentosa: genes and disease
mechanisms. Curr Genomics 2011;12:238-49. Crossref
3. National Center for Advancing Translational Sciences:
Genetic and Rare Diseases Information Center. About
GARD. Available from: http://rarediseases.info.nih.gov. Accessed 1 Jan 2021.
4. Yip SP, Cheung TS, Chu MY, et al. Novel truncating
mutations of the CHM gene in Chinese patients with
choroideremia. Mol Vis 2007;13:2183-93.
5. Lim KP, Yip SP, Cheung SC, Leung KW, Lam ST, To CH.
Novel PRPF31 and PRPH2 mutations and co-occurrence
of PRPF31 and RHO mutations in Chinese patients with
retinitis pigmentosa. Arch Ophthalmol 2009;127:784-90. Crossref