Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORTS
Mitochondrial cardiomyopathy due to
m.3243A>G mitochondrial DNA mutation
presenting in late adulthood: a case report
Elaine MC Chau, MB, BS, FRCP1; Edmond SK Ma, MD, FRCP2; Annie OO Chan, MD, FRCP1; TH Tsoi, MB, BS, FRCP1; WL Law, MS, FRCS3
1 Department of Medicine, Hong Kong Sanatorium & Hospital, Hong Kong
2 Department of Pathology, Hong Kong Sanatorium & Hospital, Hong Kong
3 Department of Surgery, Hong Kong Sanatorium & Hospital, Hong Kong
Corresponding author: Dr Elaine MC Chau (echau@hksh.com)
 Full
paper in PDF
Full
paper in PDF
Case report
Mitochondrial cardiomyopathy usually presents in
childhood and is estimated to occur in 20% to 40%
of children with mitochondrial disease. However,
it is a rare cause of cardiomyopathy in adults. We
report a case of heart failure due to hypertrophic
cardiomyopathy presenting in a 57-year-old female
patient with intestinal pseudo-obstruction and
bilateral sensorineural deafness. The constellation
of diseased organs led to suspicion of underlying
mitochondrial disease, subsequently confirmed by
genetic mitochondrial DNA (mtDNA) testing that
revealed the m.3243 A>G pathogenic variant in the
MT-TL1 gene.
A 57-year-old female was referred for
investigation of recent-onset congestive heart failure
with cardiomegaly and bilateral pleural effusions
on chest X-ray together with an elevated serum
N-terminal pro-brain natriuretic peptide level of
241 pg/mL (normal <100 pg/mL). She presented with
increasing abdominal distension and dyspnoea for
4 months. She had colonic pseudo-obstruction that
did not resolve following repeated decompression and
was referred for cardiac assessment with reference
to fitness for colectomy under general anaesthesia.
Apart from gradual hearing loss over the previous
8 years, she had been previously well. She suffered
a miscarriage at age 23 years but subsequently gave
birth to two apparently healthy daughters at age 32
and 36 years. Her main complaint over the previous
year was constipation. There was no history of
diabetes, muscle weakness, or visual problems. The
older daughter was reported to have a squint since
childhood, suggestive of ophthalmoplegia. There
was no family history of any hearing, neurological,
gastrointestinal or cardiac problems.
Clinically the patient was in distress and pain
due to the grossly dilated abdomen from the colonic
pseudo-obstruction (Fig 1). Positron emission
tomography–computed tomography scan showed
dilated bowel (up to 10 cm in diameter) from the
caecum to the anus but no evidence of malignancy. Electrocardiogram showed sinus rhythm with left
ventricular hypertrophy by voltage criteria and strain
pattern. Echocardiogram showed left ventricular
hypertrophy with mildly impaired left ventricular
systolic function (ejection fraction 40%), diastolic
dysfunction and a circumferential pericardial effusion
(Fig 2). Computed tomography coronary angiogram
was normal. Magnetic resonance imaging (MRI)
heart revealed increased T1 mapping but no late
gadolinium enhancement. Screening for amyloidosis
was negative. Neurological assessment revealed no
evidence of muscle weakness and the creatine kinase
level was normal. Computed tomography brain
showed dense calcification in bilateral lentiform
nuclei and MRI brain was essentially normal.
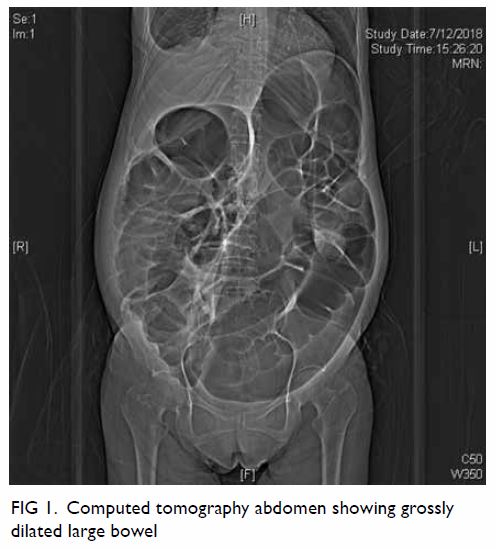
Figure 1. Computed tomography abdomen showing grossly dilated large bowel
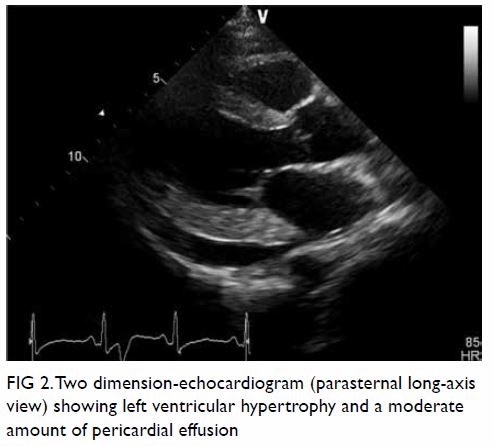
Figure 2. Two dimension-echocardiogram (parasternal long-axis view) showing left ventricular hypertrophy and a moderate amount of pericardial effusion
In view of pending rupture of the dilated colon
(up to 10 cm), she underwent subtotal colectomy
uneventfully. Histopathology of the resected colon showed no evidence of dysplasia or neoplasia. Blood
was sent for mitochondrial disorder testing and
showed a m.3243A>G mutation in the MT-TL1 gene,
with approximately 10% heteroplasmy. The resected
large bowel was micro-dissected for heteroplasmy
determination of m.3243A>G by droplet digital
polymerase chain reaction. The heteroplasmy levels
for smooth muscle, epithelium and connective
tissue of the large bowel were 77%, 66% and 46%,
respectively.
Discussion
Mitochondrial DNA disease is a multi-system
disorder that affects tissues with high energy
demands such as the heart, brain, muscle, and
endocrine system. It is caused by mutations in either
mtDNA or nuclear DNA genes. Defects in mtDNA
can be either point mutations or re-arrangements
such as deletions or duplications. The m.3243A>G
variant within the MT-TL1 gene (encoding the
mitochondrial transfer RNA) is a point mutation and
the most common heteroplasmic mtDNA disease
genotype. This mutation accounts for approximately
80% of the MELAS phenotype (mitochondrial
myopathy, encephalopathy, lactic acidosis, plus
stroke-like episodes) that features mitochondrial
encephalomyopathy, lactic acidosis and stroke-like
episodes, and which this patient did not have.
Other phenotypic variations associated with the
m.3243A>G mutation include maternally inherited
diabetes and deafness, ocular (eg, progressive
external ophthalmoplegia), gastrointestinal, cardiac,
and renal involvement. Most carriers of this
mutation are asymptomatic due to mitochondrial
heteroplasmy, implying the existence of two or
more genomes within the same cell. Usually, the proportion of heteroplasmy needs to exceed a
threshold level of 60% to 90% for the organ to be
clinically affected. The onset and extent of clinical
disease are determined by the mtDNA mutation load
and threshold. In m.3243A>G mutation carriers,
hypertrophic cardiomyopathy is found in about 18%
of patients.1
In a small study of 10 m.3243A>G mutation
carriers, the skeletal muscle mutation load
appeared to correlate with the measures of early
cardiac dysfunction on MRI, namely the torsion-to-endocardial strain ratio and radial thickening.2
Different patterns of cardiac involvement are
associated with different mtDNA mutations.
Although hypertrophic cardiomyopathy is frequently
associated with mt-tRNA (transfer RNA) mutations
such as the m.3243A>G mutation, restrictive
cardiomyopathy is associated with the m.1555A>G
mitochondrial ribosomal ribonucleic acid gene
mutation. In children with Kearns-Sayre syndrome
due to single, large-scale deletions in mtDNA,
complete heart block is common.3 In our case, the
heteroplasmy level within the different structures
in the large bowel (46%-77%) was uniformly higher
than in the blood (10%). This was consistent with the
clinical presentation of pseudo-obstruction with the
large bowel being the major organ involved.
Unfortunately, there is no cure for
mitochondrial disease. Device therapy in the form of
pacemakers and cardioverter-defibrillators may be
required for heart block and ventricular arrhythmias
associated with mitochondrial cardiomyopathy. Due
to the complexity of mitochondrial disease, it is now
recommended that affected patients be assessed
using a validated semi-quantitative clinical rating
scale, the Newcastle Mitochondrial Disease Adult
Scale, to evaluate the different systems involved
and to monitor response to treatment. Regular
cardiovascular screening is recommended with
electrocardiogram and echocardiogram performed
at least every 2 years.4
Mitochondrial DNA is strictly maternally
inherited, with only rare reports of paternal
transmission. This may be due to a dilution effect
since the sperm contain only 100 copies of mtDNA
compared with 100 000 copies in the unfertilised egg
and, secondly, there is elimination of sperm mtDNA
in normal embryos. The m.3243 A>G mutation is
usually transmitted from mother to offspring with
rare reported cases of de novo mutation.5 Genetic
counselling in mitochondrial disease may be difficult
because of heteroplasmy, mitotic segregation, and
mitochondrial bottleneck phenomenon. Phenotypic
variations associated with m.3243A>G are due to a
different percentage of mutation heteroplasmy in
affected organs. It is known that an asymptomatic
mother with a subclinical heteroplasmy level can give
birth to an affected child with a higher heteroplasmy level. This is explained by the mitochondrial
bottleneck theory, whereby redistribution of
mtDNA to daughter cells during oocyte production
and amplification of the redistributed mtDNA
during oocyte development occurs. The unequal
mitotic segregation of mtDNA during cell division
and the reduction/amplification event leads to a
random shift of mtDNA mutational load between
generations. This is thought to be responsible for the
variable levels of mutation heteroplasmy observed
in affected offspring from mothers with pathogenic
mtDNA mutations.
Conclusion
Mitochondrial cardiomyopathy is a rare cause of
heart failure presenting in adulthood. Mitochondrial
disease should be suspected when other organs
or tissues are involved. Genetic testing is a
powerful diagnostic tool. Severity and prognosis of
mitochondrial disease depend on the concentration
of heteroplasmy in the affected organs. Screening
and follow-up cardiac assessment are recommended
due to the progressive nature of cardiac involvement
and risk of heart failure and arrhythmias as a cause
of death in patients with mitochondrial mutation.
Mitochondrial mutations are maternally inherited
but the unique features of mitochondrial inheritance,
namely mitotic segregation and mitochondrial
bottleneck phenomenon, make predictions of
inheritance a challenge. Genetic counselling should
be offered to the families of an affected individual.
Author contributions
Concept or design: All authors.
Acquisition of data: EMC Chau, ESK Ma.
Analysis or interpretation of data: EMC Chau.
Drafting of the manuscript: EMC Chau.
Critical revision for important intellectual content: All authors.
Acquisition of data: EMC Chau, ESK Ma.
Analysis or interpretation of data: EMC Chau.
Drafting of the manuscript: EMC Chau.
Critical revision for important intellectual content: All authors.
The authors had contributed to the manuscript, approved the
final version for publication, and take responsibility for its
accuracy and integrity.
Conflict of interest
The authors have no conflicts of interest to disclose.
Funding/support
This case report received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
The patient was treated in accordance with the Declaration of Helsinki. The patient provided informed consent for all
procedures.
References
1. Pankuweit S, Richter A. Mitochondrial disorders with
cardiac dysfunction: an under-reported aetiology with
phenotypic heterogeneity. Eur Heart J 2015;36:2894-7. Crossref
2. Hollingsworth KG, Gorman GS, Trenell MI, et al.
Cardiomyopathy is common in patients with the
mitochondrial DNA m.3243A>G mutation and correlates
with mutation load. Neuromuscul Disord 2012;22:592-6. Crossref
3. Bates MG, Bourke JP, Giordano C, d’Amati G, Turnbull DM,
Taylor RM. Cardiac involvement in mitochondrial DNA
disease: clinical spectrum, diagnosis, and management.
Eur Heart J 2012;33:3023-3. Crossref
4. Schaefer AM, Phoenix C, Elson JL, McFarland R, Chinnery
PF, Turnbull DM. Mitochondrial disease in adults: a
scale to monitor progression and treatment. Neurology
2006;66:1932-4. Crossref
5. de Laat P, Janssen MC, Alston CL, Taylor RW, Rodenburg
RJ, Smeitink JA. Three families with ‘de novo’ m.3243A>G
mutation. BBA Clin 2016;6:19-24. Crossref