© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
COMMENTARY
Posterior reversible encephalopathy syndrome as a
neuropsychiatric manifestation of systemic lupus erythematosus
CM Ho, MB, ChB, MRCP; CC Mok, MD, FRCP (Lond)
(Edin)
Department of Medicine and Geriatrics, Tuen Mun
Hospital, Tuen Mun, Hong Kong
Corresponding author: Dr CM Ho (hocm283@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Introduction
Neuropsychiatric manifestations of systemic lupus
erythematosus (SLE) are heterogeneous and complex. Posterior reversible
encephalopathy syndrome (PRES) and its association with SLE has
increasingly been recognised.1 2 3
4 In the 1999 American College of
Rheumatology nomenclature for neuropsychiatric SLE, PRES is not classified
as a distinct primary syndrome.5 We
recently encountered a patient with active SLE who developed PRES. Given
the strong association between PRES and active SLE, and the recent
evidence for an inflammatory aetiology, we believe PRES should be
re-evaluated as a possible primary neuropsychiatric SLE syndrome.
Posterior reversible encephalopathy syndrome and
systemic lupus erythematosus
In 1996, Hinchey et al6
first described PRES as a reversible brain syndrome with typical clinical
and radiological finding. Common presentations include headache, vomiting,
altered mental function, visual symptoms, and seizures.1 2 Diagnosis is
supported by classical symptoms and typical radiological feature of
bilateral posterior subcortical brain oedema on magnetic resonance imaging
(MRI).1 Classically, PRES is linked
with accelerated hypertension, renal impairment, eclampsia, pre-eclampsia,
sepsis, cytotoxic therapy, underlying autoimmune disease, and
immunosuppressive therapy.1 After
Hinchey el al6 first described PRES in 1996, strong evidence for a link
between PRES and SLE has been established. In one case series of 120
patients with PRES, 18% were diagnosed with SLE.1
Another recent study from Taiwan involving 3746 patients with SLE
described a 0.69% prevalence of PRES episodes.2
Patients with active SLE who are receiving
immunosuppressants are at risk of developing PRES. A Korean case series
reported 15 patients with SLE who developed PRES, 80% of whom had renal
insufficiency (serum creatinine ≥132.6 μmol/L) that was associated with a
129-fold increased risk of this complication. Other risk factors for PRES
included hypertension, current treatment with high-dose steroids or
cyclophosphamide, blood transfusion, hypoalbuminaemia, and high SLE
Disease Activity Index.3 Other
immunosuppressants such as calcineurin inhibitors, mycophenolate, and
rituximab have also been implicated in the development of PRES.6 7 However, the
association between PRES and medication use should be interpreted with
caution because patients receiving these drugs usually have high
background SLE disease activity. More recently, hyperlipidaemia and
lymphopenia have been described as risk factors for PRES in patients with
SLE.8
It is important to differentiate PRES from other
active neuropsychiatric manifestations of SLE because treatment of PRES
alone does not require immunosuppression. Other differential diagnoses of
neuropsychiatric symptoms of SLE such as central nervous systemic
infection, cerebrovascular events, metabolic and electrolyte disturbances,
and adverse drug reactions must be excluded. A high index of suspicion is
needed to diagnose PRES. Typical features of headache, acute hypertension,
seizures, visual symptoms, and altered mental state should be recognised.
Urgent MRI is the standard imaging study to diagnose PRES. Prompt
treatment of PRES is necessary to prevent permanent neurological damage.
Usually, PRES is completely reversible with blood pressure control and
supportive measures; immunosuppression is indicated only for treating the
underlying active SLE.
Pathophysiology of posterior reversible encephalopathy
syndrome in systemic lupus erythematosus
The pathophysiology of PRES in SLE remains unclear.
The classical understanding is that an acute rise in blood pressure
exceeds the autoregulation of cerebral circulation, leading to increased
cerebral blood flow and hyperperfusion brain injury. On the contrary, an
excessive autoregulation response may cause a focal cerebral
vasoconstriction, leading to brain ischaemia. The attribution to
hypertensive crisis may explain part of the pathophysiology. However, the
mechanism of PRES is unlikely explained by hypertension alone. Several
cases of PRES in patients with SLE with normal blood pressure have been
reported.3 6 9 Emerging
evidence has shown that PRES in patients with SLE may be influenced by
inflammatory mechanisms.10 11
Interleukin 6 plays an important role in the
inflammatory process in neuropsychiatric SLE. Previous studies had already
revealed an elevated cerebrospinal fluid interleukin 6 level in
neuropsychiatric patients with SLE.12
Recently, a study on patients with SLE with PRES found a 2.84-fold
increase in serum interleukin 6 level compared with control patients with
active SLE without PRES.11
Interleukin 6 activates the STAT-3 pathway and up-regulates the expression
of ICAM-1, VCAM-1, and endothelial nitric oxide synthase.13 These molecules activate the endothelium of blood
vessels, increasing vascular permeability. In a normal physiological
state, this mechanism allows cell migration for normal inflammatory
processes. In PRES, the endothelial dysfunction and disruption of the
blood-brain barrier predispose patients to hyperperfusion-induced
vasogenic oedema and neurological damage.
Exemplar case
A 24-year-old woman with known SLE since childhood
was admitted to Tuen Mun Hospital, Hong Kong, in February 2018 for a
serious disease flare with profound cytopenia and worsening lupus
nephritis. The patient’s SLE had been diagnosed in 2006 when she presented
with haemolytic anaemia and diffuse global lupus nephritis (International
Society of Nephrology/Renal Pathology Society class IVG). She had tested
positive for anti-nuclear antibody screening, anti-double stranded DNA,
anti-Ro, and anti-cardiolipin antibodies. The patient’s SLE had become
unstable in the past 2 years with multiple episodes of disease relapse
involving haematological and renal systems. She became glucocorticoid
dependent and had received several treatment modalities, including
intravenous immunoglobulin, mycophenolate mofetil, cyclophosphamide,
tacrolimus, and rituximab.
On presentation, the patient had severe
thrombocytopenia (38 × 109/L), haemolytic anaemia (haemoglobin
69 g/L), serositis, acute renal function deterioration, and proteinuria
(urine protein/creatinine ratio 4.38). Because her cytopenia and kidney
disease was refractory to high-dose glucocorticoid and mycophenolic acid,
another course of intravenous immunoglobulin and rituximab was given on 8
March 2018. The patient developed headache on 16 March 2018 but a computed
tomography (CT) scan of the brain was normal. Her renal function further
deteriorated, with serum creatinine level 217 μmol/L (reference range,
50-98 μmol/L). The patient’s blood pressure increased from her baseline of
130/80 mm Hg, recorded 1 day before seizure onset, to 150/100 mm Hg on the
day of seizure onset, shortly before she suddenly developed repeated
episodes of tonic-clonic convulsions. She was transferred to the intensive
care unit and treated with intravenous levetiracetam and propofol to
control the status epilepticus and labetalol to control blood pressure. A
new CT image of the brain showed a new hypo-attenuating area affecting the
cortical and subcortical regions of bilateral occipital lobe (Fig
a). An MRI image of the brain showed bilateral white-matter oedema
with posterior and subcortical predominance (Fig b), which was compatible with PRES. After 5
days, the adequate control of the patient’s seizures and blood pressure
were achieved, and she was extubated. However, this was followed by
transient confusion and visual hallucinations for the next 2 days before a
full neurological recovery. Further doses of rituximab were given to
salvage the patient’s renal disease and cytopenia. There was no recurrence
of the seizures and a complete resolution of the brain oedema on MRI image
of the brain taken 3 months later (Fig c).
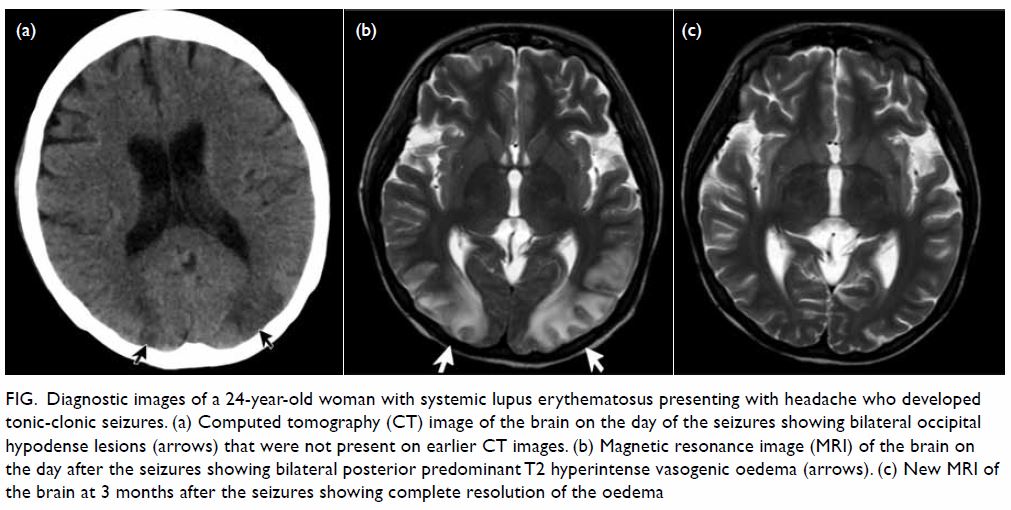
Figure. Diagnostic images of a 24-year-old woman with systemic lupus erythematosus presenting with headache who developed tonic-clonic seizures. (a) Computed tomography (CT) image of the brain on the day of the seizures showing bilateral occipital hypodense lesions (arrows) that were not present on earlier CT images. (b) Magnetic resonance image (MRI) of the brain on the day after the seizures showing bilateral posterior predominant T2 hyperintense vasogenic oedema (arrows). (c) New MRI of the brain at 3 months after the seizures showing complete resolution of the oedema
Conclusions
Prompt recognition and treatment of PRES is
important in patients with SLE. The strong association of PRES with active
SLE, particularly nephritis, and elevation of serum cytokines supports an
inflammatory process that leads to endothelial dysfunction. In this sense,
PRES should be re-evaluated as a primary neuropsychiatric manifest-ation
of SLE. The efficacy of immunosuppressive therapy in addition to blood
pressure control and other supportive measures in PRES should be further
evaluated in clinical trials.
Author contributions
CM Ho reviewed the case, extracted the patient’s
clinical information, and wrote the article. All authors performed
literature research and critical revision for the important intellectual
content. All authors had full access to the data, contributed to the
study, approved the final version for publication, and take responsibility
for its accuracy and integrity.
Conflicts of interest
The authors have no conflicts of interest to
disclose.
Funding/support
This research received no specific grant from any
funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
Informed consent was obtained from the patient.
References
1. Fugate JE, Claassen DO, Cloft HJ,
Kallmes DF, Kozak OS, Rabinstein AA. Posterior reversible encephalopathy
syndrome: associated clinical and radiologic findings. Mayo Clin Proc
2010;85:427-32. Crossref
2. Lai CC, Chen WS, Chang YS, et al.
Clinical features and outcomes of posterior reversible encephalopathy
syndrome in patients with systemic lupus erythematosus. Arthritis Care Res
(Hoboken) 2013;65:1766-74. Crossref
3. Jung SM, Moon SJ, Kwok SK, et al.
Posterior reversible encephalopathy syndrome in Korean patients with
systemic lupus erythematosus: risk factors and clinical outcome. Lupus
2013;22:885-91. Crossref
4. Damrongpipatkul U, Oranratanachai K,
Kasitanon N, Wuttiplakorn S, Louthrenoo W. Clinical features, outcome, and
associated factors for posterior reversible encephalopathy in Thai
patients with systemic lupus erythematosus: a case-control study. Clin
Rheumatol 2018;37:691-702. Crossref
5. The American College of Rheumatology
nomenclature and case definitions for neuropsychiatric lupus syndromes.
Arthritis Rheum 1999;42:599-608. Crossref
6. Hinchey J, Chaves C, Appignani B, et al.
A reversible posterior leukoencephalopathy syndrome. N Engl J Med
1996;334:494-500. Crossref
7. Mondal S, Goswami RP, Sinha D, et al.
Posterior reversible encephalopathy syndrome in a patient with lupus
nephritis on rituximab therapy: a challenge to find the offender. Lupus
2016;25:445-6. Crossref
8. Merayo-Chalico J, Apodaca E,
Barrera-Vargas A, et al. Clinical outcomes and risk factors for posterior
reversible encephalopathy syndrome in systemic lupus erythematosus: a
multicentric case-control study. J Neurol Neurosurg Psychiatry
2016;87:287-94. Crossref
9. Kur JK, Esdaile JM. Posterior reversible
encephalopathy syndrome—an underrecognized manifestation of systemic lupus
erythematosus. J Rheumatol 2006;33:2178-83.
10. Marra A, Vargas M, Striano P, Del
Guercio L, Buonanno P, Servillo G. Posterior reversible encephalopathy
syndrome: the endothelial hypotheses. Med Hypotheses 2014;82:619-22. Crossref
11. Merayo-Chalico J, Barrera-Vargas A,
Juárez-Vega G, Alcocer-Varela J, Arauz A, Gómez-Martín D. Differential
serum cytokine profile in patient with systemic lupus erythematosus and
posterior reversible encephalopathy syndrome. Clin Exp Immunol
2018;192:165-70. Crossref
12. Fragoso-Loyo H, Richaud-Patin Y,
Orozco-Narváez, et al. Interleukin-6 and chemokines in the
neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis
Rheum 2007;56:1242-50. Crossref
13. Wei Z, Jiang W, Wang H, et al. The
IL-6/STAT3 pathway regulates adhesion molecules and cytoskeleton of
endothelial cells in thromboangiitis obliterans. Cell Signal
2018;44:118-26. Crossref