Hong Kong Med J 2024;30:Epub 23 Dec 2024
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
CASE REPORT
Collapsing glomerulopathy as a rare cause of rapidly progressive renal failure in adolescence: two case reports
Yeşim Özdemir Atikel, MD1; Betül Öğüt, MD2; İpek Işık Gönül, MD2; Necla Buyan, MD1; Sevcan A Bakkaloğlu, MD1
1 Department of Pediatric Nephrology, Gazi University Faculty of Medicine, Gazi University, Ankara, Turkey
2 Department of Pathology, Gazi University Faculty of Medicine, Gazi University, Ankara, Turkey
Corresponding author: Prof Yeşim Özdemir Atikel (yesozdemir@gmail.com)
 Full paper in PDF
Full paper in PDF
Case presentations
Case 1
A 17-year-old male was referred to our institution in
January 2016 due to elevated serum creatinine level
of 1.85 mg/dL and nephrotic proteinuria level of 6839
mg/day. He had a history of epilepsy and had used
various antiepileptic drugs (phenobarbital, valproic
acid, and carbamazepine) from the ages 3 to 14
years. Physical examination revealed lower extremity
oedema and a blood pressure of 140/90 mm Hg.
Laboratory tests on admission showed a blood urea
nitrogen level of 24 mg/dL, serum creatinine level of
1.68 mg/dL and a serum albumin level of 2.7 g/dL.
Urine microscopy revealed three red blood cells
per high-power field. A 24-hour urine collection
revealed massive proteinuria level of 10.957 mg/day
(296 mg/m2/h). Serum complement levels were
normal and autoimmune tests (antinuclear
antibodies, anti–double-stranded DNA antibodies,
anti–glomerular basal membrane antibodies, and
anti-neutrophil cytoplasmic antibodies) were negative. Viral serology, including hepatitis B virus,
hepatitis C virus, human immunodeficiency virus,
Epstein–Barr virus, cytomegalovirus, and parvovirus,
was also negative. Abdominal ultrasound revealed
increased echogenicity in the renal parenchyma.
Treatment with enalapril at a dose of
0.4 mg/kg/day was started. Kidney biopsy was
performed on the seventh day after admission and
showed compatibility with collapsing glomerulopathy
(CG) [Fig 1]. Methylprednisolone boluses of 1 g were
administered for 5 consecutive days, followed by
oral prednisone at a dose of 60 mg/m2/day. By the
15th day, serum creatinine levels were at 2.1 mg/dL,
serum albumin level at 1.9 g/dL, and 24-hour urine
protein level at 17.8 g/day. Mycophenolate mofetil
was added to the treatment regimen. On the 40th day,
serum creatinine level had increased to 4.2 mg/dL
with proteinuria of 13.3 g/day, leading to the initiation
of rituximab and tapering of prednisolone. By the
60th day, mycophenolate mofetil was discontinued
due to leukopenia; however, the patient had
completed the 4 doses of weekly 375 mg/m2/dose rituximab treatment. Additionally, he received
albumin infusions, diuretics, and antihypertensives.
Since the clinical features and laboratory parameters
did not improve, the patient underwent plasma
exchange. After two sessions, haemodialysis was
required due to worsening symptoms, uncontrolled
hypervolaemia, and renal failure. No additional
immunosuppressive was given at that time and the
patient continued to receive haemodialysis. Genetic
testing for mutations of the NPHS1 and NPHS2
genes were negative.
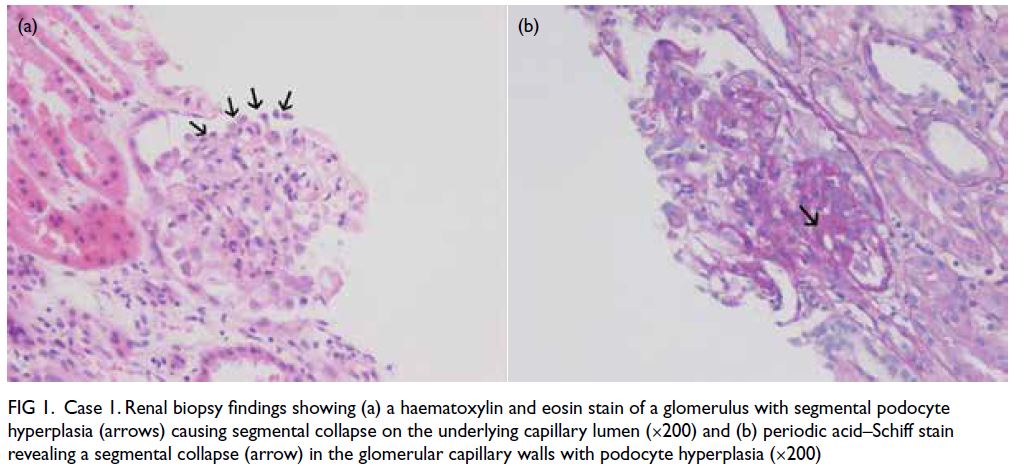
Figure 1. Case 1. Renal biopsy findings showing (a) a haematoxylin and eosin stain of a glomerulus with segmental podocyte hyperplasia (arrows) causing segmental collapse on the underlying capillary lumen (×200) and (b) periodic acid–Schiff stain revealing a segmental collapse (arrow) in the glomerular capillary walls with podocyte hyperplasia (×200)
Case 2
Another 17-year-old male was admitted to our
institution in February 2016 for syncope. He had
a history of headaches with intermittent vomiting
for the previous 2 months and had been treated
with metamizole, domperidone, zolmitriptan, and
diclofenac. His mother had a history of minimal
change disease aged 6 years. The patient’s blood
pressure was measured as 200/120 mm Hg, and
hypertensive retinopathy was observed during
the ophthalmological examination. Initial serum
creatinine level was 4.7 mg/dL and serum albumin
level was 3.4 g/dL. Ferritin and parathyroid hormone
levels were 274 ng/mL and 220 pg/mL, respectively.
Microscopic urinalysis showed eight red blood cells
per high-power field. He had nephrotic proteinuria
of 3820 mg/day (91.5 mg/m2/h). Viral serology
and autoimmune tests (antinuclear antibodies,
anti–double-stranded DNA antibodies, anti–glomerular basal membrane antibodies, and anti-neutrophil
cytoplasmic antibodies) were negative
and complement levels were normal. Abdominal
ultrasound revealed increased renal echogenicity.
Cranial magnetic resonance imaging showed signs
of posterior reversible encephalopathy syndrome. Hypertension was controlled using intravenous
and oral antihypertensives (esmolol, captopril,
amlodipine, doxazosin, and minoxidil). On the
fourth day, the serum creatinine level increased
to 5.9 mg/dL and the albumin level decreased to
2.4 g/dL. Kidney biopsy showed severe CG (Fig 2). Because the findings were chronic, no steroids
or other immunosuppressive treatment were
administered. Genetic testing for mutations of
the NPHS1 and NPHS2 genes was negative. By the
fifth month, the patient’s serum creatinine level
had reached 6.9 mg/dL. After 1 year of peritoneal
dialysis, he received a renal transplant.
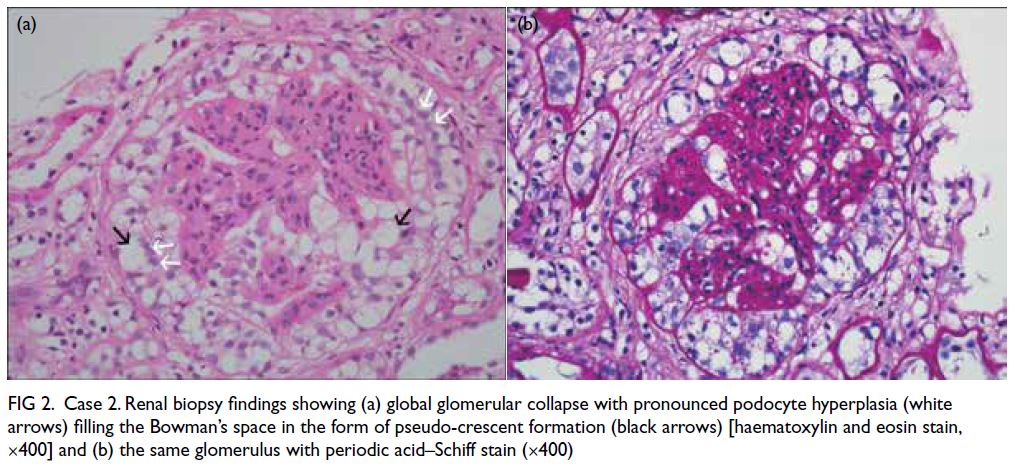
Figure 2. Case 2. Renal biopsy findings showing (a) global glomerular collapse with pronounced podocyte hyperplasia (white arrows) filling the Bowman’s space in the form of pseudo-crescent formation (black arrows) [haematoxylin and eosin stain, ×400] and (b) the same glomerulus with periodic acid–Schiff stain (×400)
Discussion
Collapsing glomerulopathy is a histopathological
pattern of podocytopathies.1 It was previously
classified as a variant of focal segmental
glomerulosclerosis (FSGS), known as collapsing
FSGS.2 3 4 However, it is more severe at the initial stage
and progresses more rapidly to end-stage kidney
disease compared with non-collapsing FSGS, even
when treatment is given.2 3 4 5 It typically presents with
nephrotic proteinuria and elevated serum creatinine
level, and is rare among children.3 5
Both patients had high serum creatinine
level, nephrotic proteinuria, and hypertension. To
establish the exact diagnosis and determine the
prognosis, a kidney biopsy was performed as the gold
standard for diagnosis. Histopathological findings
of CG include glomerular capillary collapse in at
least one glomerulus; hyperplasia and hypertrophy
of visceral epithelial cells leading to pseudo-crescent
formation; presence of periodic acid–Schiff-positive
hyaline droplets in visceral epithelial cell cytoplasm;
and severe tubulointerstitial inflammation in the
early stages. Glomerulosclerosis and interstitial fibrosis are observed in the late stages, and
immunofluorescence assay is typically negative.1 2 3 4 6
Kidney biopsies in both cases showed advanced
CG with global glomerulosclerosis and interstitial
fibrosis (Figs 1 and 2).
Collapsing glomerulopathy can be either
idiopathic (primary), genetic (familial), or reactive
(secondary).1 The idiopathic form is characterised by
the loss of maturity markers and the re-expression
of immaturity markers leading to the proliferation
of immature podocytes.1 Secondary causes of CG
include infections (human immunodeficiency virus,
parvovirus B19, cytomegalovirus, hepatitis C virus,
severe acute respiratory syndrome coronavirus
2), drugs (including valproic acid and anabolic
steroids), autoimmune diseases (such as systemic
lupus erythematosus), and malignancies.1 2 3 4 7 Genetic
CG is associated with mitochondrial dysfunction
that causes podocyte proliferation.1 Case 1 had
a history of long-term use of antiepileptic drugs
(phenobarbital, valproic acid, and carbamazepine).
However, we found no other aetiological factors
in either patient. Therefore, we concluded that
while the aetiology in Case 1 could be idiopathic or
valproic acid–related, it was idiopathic in Case 2.
There is no specific treatment for CG2; as such,
the mainstay of therapy is for the disorders resulting
from nephrotic syndrome (such as hypertension and
oedema), treatment of the underlying conditions
(such as infections and autoimmune diseases),
and immunosuppressive therapy.8 Possible factors
for progression to end-stage kidney disease in CG
include a serum creatinine level >2 mg/dL at the time
of biopsy, proteinuria >8 g/day and lack of remission,
collapsing lesions in >20% of glomeruli, and the severe
tubular changes and interstitial fibrosis.3 9 10 In Case
1, the rationale for aggressive immunosuppressive
treatment was based on an initial serum creatinine
level of 1.6 mg/dL, intense polymorphonuclear
leukocytes and eosinophil infiltration, and 2 out of
24 glomeruli showing glomerulosclerosis. Case 2
did not receive immunosuppressive treatment due
to the chronicity of the disease and advanced global
glomerulosclerosis (67%). The Table summarises the
clinical findings in both patients.
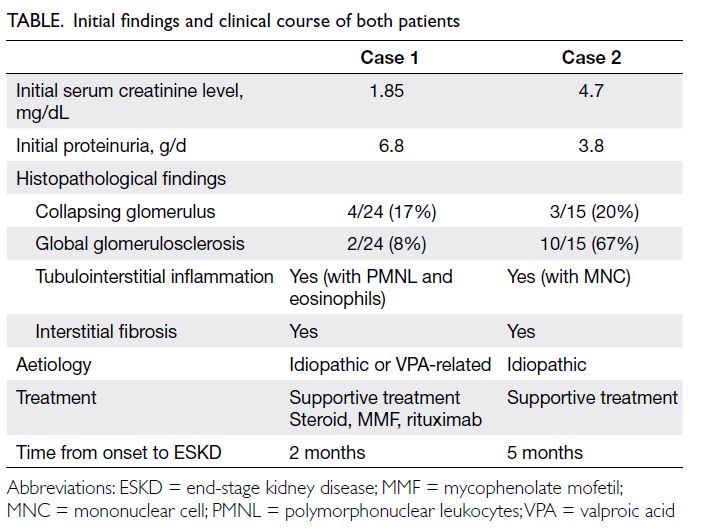
Table. Initial findings and clinical course of both patients
Conclusion
It is important to recognise that CG is a separate
clinicopathological entity from FSGS. Due to the
poor response to immunosuppressive drugs and the
potential for renal transplantation, we recommend
avoiding aggressive immunosuppressive therapy for
patients with poor prognostic factors at the time of
diagnosis. This approach helps minimise the side-effects
of cumulative immunosuppression.
Author contributions
Concept or design: All authors.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: Y Özdemir Atikel, SA Bakkaloğlu.
Critical revision of the manuscript for important intellectual content: All authors.
Acquisition of data: All authors.
Analysis or interpretation of data: All authors.
Drafting of the manuscript: Y Özdemir Atikel, SA Bakkaloğlu.
Critical revision of the manuscript for important intellectual content: All authors.
All authors had full access to the data, contributed to the study, approved the final version for publication, and take responsibility for its accuracy and integrity.
Conflicts of interest
All authors have disclosed no conflicts of interest.
Declaration
The two cases have been presented during oral presentations
at the IPNA Teaching Course of the 5th Southeastern Europe
Pediatric Nephrology Working Group Meeting inSkopje,
Macedonia, 10-11 June 2016.
Funding/support
This study received no specific grant from any funding agency
in the public, commercial, or not-for-profit sectors.
Ethics approval
Both patients were treated in accordance with the Declaration
of Helsinki. Written informed consent for publication was
obtained from both patients and their parents.
References
1. Barisoni L, Schnaper HW, Kopp JB. A proposed taxonomy
for the podocytopathies: a reassessment of the primary
nephrotic diseases. Clin J Am Soc Nephrol 2007;2:529-42. Crossref
2. Albaqumi M, Soos TJ, Barisoni L, Nelson PJ. Collapsing
glomerulopathy. J Am Soc Nephrol 2006;17:2854-63. Crossref
3. Mubarak M. Collapsing focal segmental glomerulosclerosis:
current concepts. World J Nephrol 2012;1:35-42. Crossref
4. Ferreira AC, Carvalho D, Carvalho F, Galvão MJ, Nolasco F. Collapsing glomerulopathy in Portugal: a review of the
histological and clinical findings in HIV and non-HIV
patients. Nephrol Dial Transplant 2011;26:2209-15. Crossref
5. Gulati A, Sharma A, Hari P, Dinda AK, Bagga A. Idiopathic
collapsing glomerulopathy in children. Clin Exp Nephrol
2008;12:348-53. Crossref
6. Fogo AB, Lusco MA, Najafian B, Alpers CE. AJKD Atlas of
Renal Pathology: collapsing glomerulopathy. Am J Kidney
Dis 2015;66:e3-4. Crossref
7. Nasr SH, Kopp JB. COVID-19–associated collapsing
glomerulopathy: an emerging entity. Kidney Int Rep 2020;5:759-61. Crossref
8. Cutrim ÉM, Neves PD, Campos MA, et al. Collapsing
glomerulopathy: a review by the Collapsing Brazilian
Consortium. Front Med (Lausanne) 2022;9:846173. Crossref
9. Laurinavicius A, Hurwitz S, Rennke HG. Collapsing
glomerulopathy in HIV and non-HIV patients: a
clinicopathological and follow-up study. Kidney Int
1999;56:2203-13. Crossref
10. Valeri A, Barisoni L, Appel GB, Seigle R, D’Agati V.
Idiopathic collapsing focal segmental glomerulosclerosis: a
clinicopathologic study. Kidney Int 1996;50:1734-46. Crossref