Hong Kong Med J 2019 Dec;25(6):444–52 | Epub 4 Dec 2019
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
Natural clinical course of progressive supranuclear
palsy in Chinese patients in Hong Kong
YF Shea, FHKAM (Medicine), FHKCP1; Alex
CK Shum, FHKAM (Medicine), FHKCP2; SC Lee, BHS (Nursing)1;
Patrick KC Chiu, FHKAM (Medicine), FHKCP1; KS Leung, FHKAM
(Medicine), FHKCP2; YK Kwan, FHKAM (Medicine), FHKCP2;
Francis CK Mok, FHKAM (Medicine), FHKCP2; Felix HW Chan, FHKAM
(Medicine), FHKCP2
1 Department of Medicine, Queen Mary
Hospital, Pokfulam, Hong Kong
2 Department of Medicine and Geriatrics,
Tuen Mun Hospital, Tuen Mun, Hong Kong
Corresponding author: Dr YF Shea (elphashea@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
Introduction: Progressive
supranuclear palsy (PSP) is a common type of atypical parkinsonism. To
the best of our knowledge, there has been no study of its natural
clinical course among Chinese patients.
Methods: This retrospective
study included 21 patients with PSP who had radiological evidence of
midbrain atrophy (confirmed by magnetic resonance imaging) from the
geriatrics clinics of Queen Mary Hospital and Tuen Mun Hospital.
Clinical information was retrieved from clinical records, including age
at onset, age at presentation, age at death, duration of symptoms, level
of education, sex, presenting scores on Cantonese version of Mini-Mental
State Examination, clinical symptoms, and history of levodopa or
dopamine agonist intake and response. Clinical symptoms were clustered
into the following categories and the dates of development of these
symptoms were determined: motor symptoms, bulbar symptoms, cognitive
symptoms, and others.
Results: Motor symptoms
developed early in the clinical course of disease. Cox proportional
hazards modelling showed that the number of episodes of pneumonia, time
to vertical gaze palsy, and presence of pneumonia were predictive of
mortality. Apathy, dysphagia, pneumonia, caregiver stress, and pressure
injuries were predictive of mortality when analysed as time-dependent
covariates. There was a significant negative correlation between the age
at presentation and time to mortality from presentation (Pearson
correlation=-0.54, P=0.04). Approximately 40% of caregivers complained
of stress during the clinical course of disease.
Conclusion: Important clinical
milestones, including the development of dysphagia, vertical gaze palsy,
significant caregiver stress, pressure injuries, and pneumonia, may
guide advanced care planning for patients with PSP.
New knowledge added by this study
- Although this was a small cohort, 57% of patients with progressive supranuclear palsy (PSP) were initially misdiagnosed.
- Important clinical milestones, including the development of apathy, dysphagia, vertical gaze palsy, significant caregiver stress, pressure injuries, and pneumonia, were predictive of mortality in patients with PSP.
- Monitoring of vertical gaze palsy or levodopa response is important throughout the clinical course of disease in patients with PSP.
- Important clinical milestones, including the development of dysphagia, vertical gaze palsy, significant caregiver stress, pressure injuries and pneumonia, may be used to guide the ideal timing for discussions of advanced care planning for patients with PSP.
Introduction
With the ageing of the Hong Kong population,
clinicians must monitor increasing numbers of patients with
neurodegenerative disease. Progressive supranuclear palsy (PSP) is a
common form of atypical parkinsonism,1
with a prevalence of up to 18 cases per 100 000 people.1 Pathologically, PSP is characterised by the presence of
neurofibrillary tangles, neuropil threads, or both in the basal ganglia
and brainstem.1 In patients with
classical Richardson’s syndrome, the disease is characterised by early
postural instability, falls, vertical gaze palsy, parkinsonism with poor
response to levodopa, pseudobulbar palsy, and frontal release signs.2 Increasingly, patients with PSP have been reported to
exhibit the following manifestations: parkinsonism, progressive gait
freezing, corticobasal syndrome, apraxia of speech, frontal presentation,
or cerebellar ataxia.2 Despite
advancements in understanding the disease, there remains no approved
treatment for PSP.
In addition to the need for accurate diagnosis of
PSP, the natural clinical course of the disease is a concern for
caregivers.3 4 Important problems for patients with PSP include
increased risks of falls, dysphagia, aspiration pneumonia, pressure
injuries, caregiver stress leading to institutionalisation, and long-term
mortality.1 Given the lack of
definitive treatment, it remains prudent for clinicians to educate
caregivers regarding the natural course of PSP, which will ensure that
caregivers are better prepared to care for their relatives; it will also
allow implementation of different methods to avoid long-term
complications, and may facilitate discussions of advanced care planning
(ACP) at earlier stages of disease.3
In particular, clinicians need to identify the appropriate timing to
discuss ACP. Previous studies have shown that the mean age at onset of PSP
is 61 to 67.2 years, and that the disease affects both sexes equally;
moreover, the median survival ranges from 5.3 to 10.2 years.5 Factors predictive of mortality included age at onset,
early clinical milestones (eg, falls, vertical gaze palsy, neck or limb
stiffness, dysphagia, and incontinence), cognitive impairment, language
impairment, autonomic dysfunction, male sex, and certain subtypes of PSP,
such as classical Richardson’s syndrome and pneumonia.6 7 8 9 10 11 12 13 14 15 16
To the best of our knowledge, there have been no
studies of the natural clinical course of disease in Chinese patients with
PSP. This information is important for clinical treatment and the design
of future intervention studies (eg, for the purposes of sample size
estimation). In the present study, we hypothesised that bulbar symptoms
and pneumonia could predict mortality in patients with PSP. The aims of
this study were (1) to calculate the prevalences of motor symptoms,
cognitive symptoms, bulbar symptoms, other systemic symptoms, and
long-term outcomes (eg, falls, tube feeding, pressure injuries, and
institutionalisation) during the clinical course of PSP, and (2) to
identify factors predictive of mortality among patients with PSP.
Methods
Patients
This retrospective study protocol was approved by
the Institutional Review Board of the University of Hong Kong/Hospital
Authority Hong Kong West Cluster (HKU/HA HKW IRB; Approval No. UW 17-483)
and New Territories West Cluster Clinical and Research Ethics Committee
(NTWC CREC; Approval No. NTWC/CREC/17127); the requirement for informed
consent was waived by the review board. This study comprised a
retrospective review of the clinical records of all patients who presented
to the geriatrics clinics of Queen Mary Hospital and Tuen Mun Hospital
between 1 January 2008 and 30 December 2017. All patients had at least 1
year of clinical follow-up and fulfilled the latest Movement Disorder
Society Criteria for clinical diagnosis of PSP.2
In addition, magnetic resonance imaging scans showed radiological evidence
of midbrain atrophy (Hummingbird sign or Morning Glory sign) in all
patients, according to radiological reports prepared by licensed
radiologists.1 Twenty-five patients
with clinically probable PSP and radiological evidence of midbrain atrophy
were considered for inclusion in this study. Four patients were excluded
because their clinical history and radiology findings were not suggestive
of PSP. Finally, 21 patients were included: 19 had Richardson syndrome
variant, one had PSP-corticobasal syndrome, and one had PSP with language
impairment. The patient who had PSP with language impairment was
previously described.17 Seven and
14 patients were recruited from Queen Mary Hospital and Tuen Mun Hospital,
respectively.
Baseline clinical information retrieved
Clinical information was retrospectively retrieved
from clinical records, including age at onset, age at presentation, age at
death, duration of symptoms, level of education, sex, presenting scores on
Cantonese version of Mini-Mental State Examination (C-MMSE),18 clinical symptoms, and history of levodopa or
dopamine agonists intake or response. Clinical symptoms were clustered
into the following categories: motor symptoms (including limb or neck
stiffness, slowness of movement, balance impairment, gait impairment,
falls, tremor, and vertical gaze palsy), bulbar symptoms (including
dysarthria, dysphagia, and drooling), cognitive symptoms (including memory
impairment, apathy, apraxia, dysexecutive syndrome, behavioural
disinhibition, repetitive motor behaviour, hyperorality, and visual
hallucination), and others (including faecal or urinary incontinence,
constipation, insomnia, depression, and caregiver stress).6 The dates of development of the above symptom clusters
were retrospectively determined.
‘Age at presentation’ was defined as the age at
which the patient first presented to the geriatrics clinic. ‘Duration of
symptoms’ was defined as the time between the first appearance of clinical
symptoms of neurodegenerative disease and the first presentation. ‘Age at
onset’ was defined as the difference between the ‘age at presentation’ and
the ‘duration of symptoms’. ‘Disease duration’ was defined as the
difference between ‘age at onset’ and ‘age at death’ or the last date of
follow-up. ‘Time to diagnosis’ was defined as the time between the date of
disease onset and the date of diagnosis with PSP. The times to development
of the above categories of symptoms were calculated in relation to both
disease onset and presentation.
Long-term outcomes
Long-term outcomes were recorded, including falls,
dysphagia, pneumonia, pressure sore development, and mortality. ‘Time to
event’ was defined by the difference between the onset of clinical
symptoms and first appearance of these long-term events. The time to each
event from the time of first presentation was also calculated.
Falls
These were defined as events that resulted in the
patient’s body or body part inadvertently coming to rest on the ground or
other surface lower than the body. The dates and numbers of falls were
recorded. Geriatric day hospital training was recorded, including the
pre-/post-training elderly mobility scale.19
Parkinsonism medications were often titrated in the geriatric day
hospital.
Pneumonia
A diagnosis of pneumonia was made based on the
following criteria: clinical signs and symptoms, white cell count of
≥10×109/L or proportion of neutrophils of ≥80%, fever (body
temperature of ≥37.6℃), and new infiltrates or consolidations on chest
radiography (X-ray or computed tomography).20
The dates and total numbers of pneumonia diagnoses were recorded.
Dysphagia
Any documentation of dysphagia by a speech
therapist was recorded; alternatively (if available), reports of video
fluoroscopic swallowing studies were obtained. Penetration was defined as
the entry of barium material into the airway without passage below the
vocal cords; aspiration was defined as the passage of barium material
below the level of the vocal cords.21
The dates of diagnosis of dysphagia and tube feeding were recorded.
Pressure injuries
The locations of pressure injuries and their
stages, according to National Pressure Ulcer Advisory Panel guidelines,
were recorded.22 The dates of
discovery of pressure injuries were recorded.
Institutionalisation
This was defined as institutionalisation of the
patient, regardless of the level of care (eg, personal care facility or
health care facility). The dates of institutionalisation were recorded
where possible.
Mortality
The date and cause of death were recorded.
Statistical analysis
For descriptive statistics, continuous variables
with normal distributions were expressed as means ± standard deviations;
variables that did not exhibit a normal distribution were expressed as
medians (interquartile ranges). Symptom prevalences (cumulative
incidences) were estimated using Kaplan-Meier method, with the first
presentation defined as time zero. Patients who did not exhibit a
particular symptom by the most recent assessment were censored at that
point. Median times to clinical events were used as cut-offs to define
‘early’ or ‘late’ development of those events (binary classification).
Time zero was consistently defined as the first clinical presentation or
onset of disease, while the event time was defined as the number of months
from first presentation or disease onset to occurrence of the event. Cox
proportional hazards modelling was used to identify factors predictive of
mortality based on the above binary classification or clinical events as
time-dependent covariates. Pearson correlation coefficients were used to
study the correlations between age at presentation and time to mortality
(from the date of presentation). A two-tailed P value of <0.05 was
considered statistically significant. All statistical analyses were
performed using SPSS for Windows (version 24; IBM Corp, Armonk [NY],
United States).
Results
Basic demographics
Twenty-one patients were included in this analysis,
with a total of 1671.4 months of follow-up from onset (1428.4 months from
presentation). The baseline demographics are summarised in Table
1. The mean age of the patients at presentation was 67.6 ± 9.4
years, and most patients were men (76.2%). Fifty-seven percent of the
patients received another diagnosis at the time of presentation: nine
patients were diagnosed with Parkinson’s disease, one patient was
diagnosed with Lewy body dementia, one patient was diagnosed with cervical
myelopathy, and one patient was diagnosed with myasthenia gravis. None of
the patients showed improvement when treated with levodopa or a dopamine
agonist. Six patients (28.6%) exhibited dementia at the time of
presentation. Seventeen patients (81%) had been referred to the geriatric
day hospital for rehabilitation after a median of 20 months from
presentation; they showed improvement in elderly mobility scale score
(pre-geriatric day hospital elderly mobility scale score vs post-geriatric
day hospital elderly mobility scale score: 14 ± 4.6 vs 16 ± 4.3,
respectively, P=0.02). Fifteen patients (71.4%) died during follow-up with
mean survival of 6.1 ± 2.6 years from onset (5.2 ± 3.2 years from
presentation); 12 of these 15 patients (80%) died of pneumonia, while two
(13.3%) died of sudden cardiac arrest.
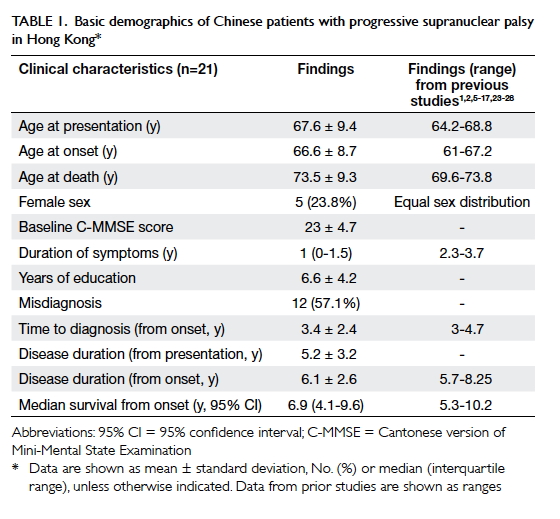
Table 1. Basic demographics of Chinese patients with progressive supranuclear palsy in Hong Kong
Clinical features
Among the categories of potential symptoms, motor
symptoms were most prevalent during initial presentation (specific
symptoms affected up to 33.3% of patients) [Fig 1]. Motor manifestations were among the earliest
clinical features observed in patients with PSP (online supplementary Appendix). The most frequent
motor symptoms at the time of presentation were limb stiffness (33.3%) and
gait impairment (28.6%). Vertical gaze palsy was present in 19% of
patients at the time of presentation, but eventually affected all patients
(100%). The prevalence of gait impairment increased rapidly, such that
≥80% of the patients were affected within 3 years after presentation. All
motor features showed increased in prevalence over time, with final
prevalences ranging from 47.6% to 100%. Regarding bulbar symptoms,
dysarthria was the most frequent presenting symptom (9.5%). Both
dysarthria and dysphagia reached 100% prevalence over time (Fig
1).
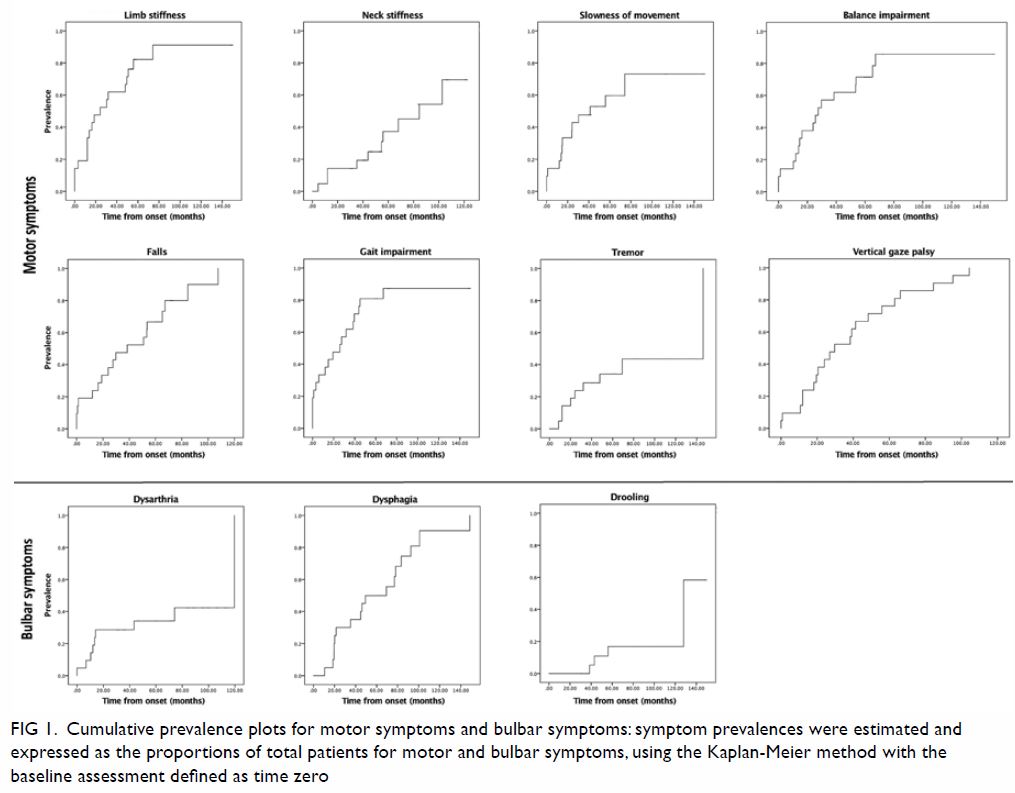
Figure 1. Cumulative prevalence plots for motor symptoms and bulbar symptoms: symptom prevalences were estimated and expressed as the proportions of total patients for motor and bulbar symptoms, using the Kaplan-Meier method with the baseline assessment defined as time zero
Regarding cognitive symptoms, memory impairment and
apathy were the two most frequent presenting symptoms, with prevalences of
33.3% and 23.8%, respectively (Fig 2). The respective prevalences of memory
impairment and apathy increased to >65% and >45% over time. The
prevalence of dysexecutive syndrome reached 28.6% during the clinical
course of disease. Other cognitive symptoms relevant to patients with PSP
showed lower prevalence, including apraxia, behavioural disinhibition,
repetitive motor behaviour, hyperorality, and visual hallucination; the
highest prevalence for any of these symptoms was 9.5% throughout the
clinical course of disease. The degree of caregiver stress also increased
with progression of disease, such that it reached approximately 40% within
5 years after initial presentation.
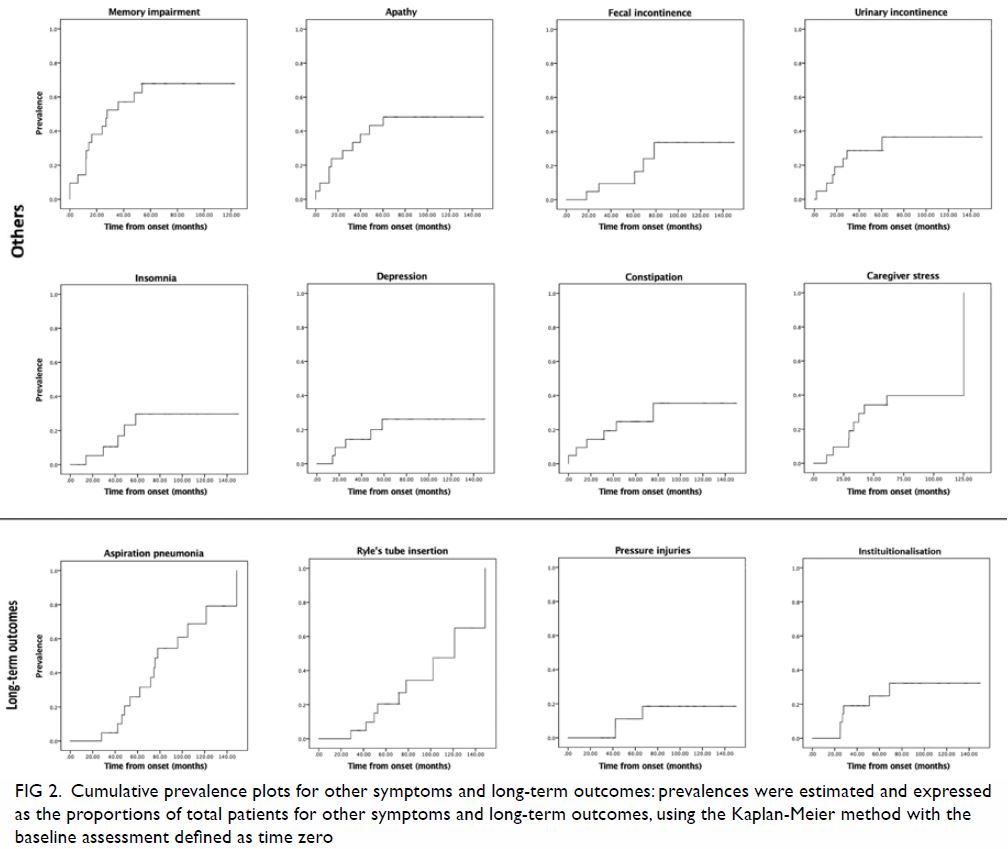
Figure 2. Cumulative prevalence plots for other symptoms and long-term outcomes: prevalences were estimated and expressed as the proportions of total patients for other symptoms and long-term outcomes, using the Kaplan-Meier method with the baseline assessment defined as time zero
Regarding systemic symptoms, faecal and urinary
incontinence showed the highest prevalences (both reached approximately
40%), particularly in the later stages of disease (Fig 2). Regarding long-term outcomes, the
prevalences of aspiration pneumonia and dysphagia requiring Ryle’s tube
insertion both reached 100% over time (Fig 2).
Factors predicting mortality
Using median time to clinical events as cut-off
(binary classification) [online supplementary Appendix] and with analysis in
a Cox proportional hazards model, our results showed that earlier
development of vertical gaze palsy (hazard ratio [HR]=4.4, 95% confidence
interval [CI]=1.4-13.9, P=0.01) and earlier development of pneumonia
(HR=10.9, 95% CI=2.2-53.3, P=0.003) were predictive of mortality from
disease onset (Table 2). Multivariate analysis showed that earlier
development of vertical gaze palsy (HR=3.8, 95% CI=1.1-13.0, P=0.04) and
earlier development of pneumonia (HR=10.4, 95% CI=1.9-55.7, P=0.006) were
predictive of mortality from disease onset (Table 2). Earlier development of vertical gaze palsy
(HR=3.3, 95% CI=1.1-10.5, P=0.01), earlier development of dysphagia
(HR=3.7, 95% CI=1.0-12.9, P=0.04), and earlier development of pneumonia
(HR=10.6, 95% CI=2.2-52.0, P=0.004) were predictive of mortality from
disease presentation (Table 2). Multivariate analysis showed that only
earlier development of pneumonia (HR=9.9, 95% CI=1.9-51.8, P=0.007) was
predictive of mortality from presentation (Table 2). The number of episodes of pneumonia was
also predictive of mortality in patients with PSP, indicating that
pneumonia is a major cause of mortality (Table 2). There was a significant negative
correlation between the age at presentation and time to mortality from
presentation (Pearson correlation=-0.54, P=0.04).
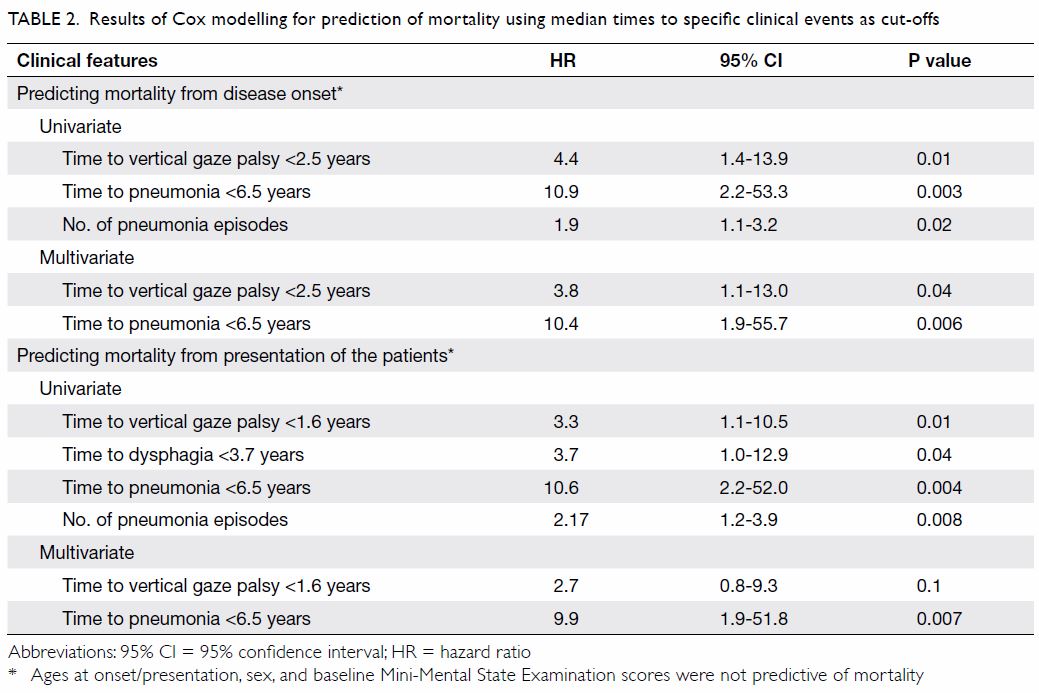
Table 2. Results of Cox modelling for prediction of mortality using median times to specific clinical events as cut-offs
Using clinical events as time-dependent covariates
in Cox modelling for prediction of mortality, we found that apathy,
dysphagia, Ryle’s tube feeding, pneumonia, and pressure injuries were
predictive of mortality from both disease onset and presentation (Table
3). Caregiver stress was only predictive of mortality from
presentation (Table 3).
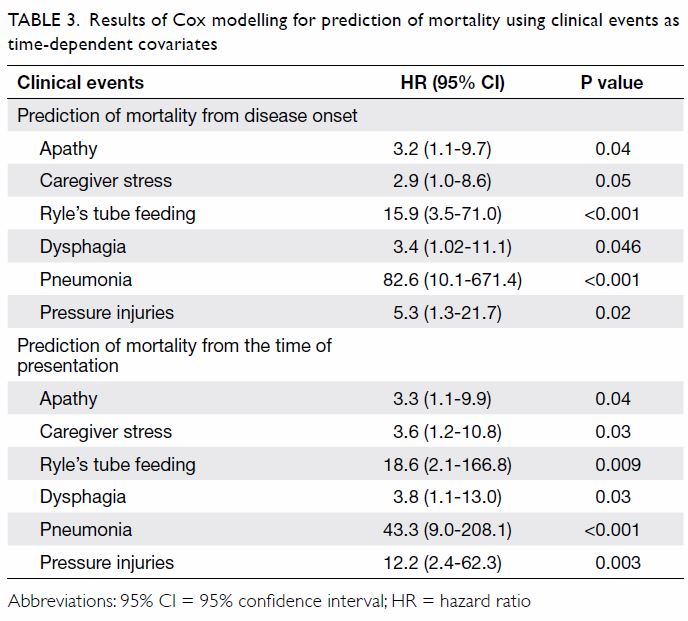
Table 3. Results of Cox modelling for prediction of mortality using clinical events as time-dependent covariates
Discussion
An accurate diagnosis of PSP is important for
management of the disease in affected patients. However, only 43% of the
patients in this study received a correct diagnosis at the time of initial
presentation. This is potentially because vertical gaze palsy was not
present initially and only developed during clinical follow-up (median
time to develop, 19.6 months; online supplementary Appendix). In addition, 43% of
patients were initially misdiagnosed with Parkinson’s disease; this group
of patients may have had PSP with parkinsonism.2
Clinicians should regularly assess patients with parkinsonism for the
presence of any vertical gaze palsy or poor response to levodopa, in order
to correctly identify patients with PSP. Our reported mean time to
diagnosis of 3 years was similar to the duration reported in previous
studies (mean, 3.1-4 years).1 2 5 6 7 8 9 10 11 12 13 14 15 16 17 23 24 25 26 27 28
With regard to clinical features, our cohort of
patients exhibited early development of motor symptoms, which was
consistent with previous studies.1
2 5
6 7
8 9
10 11
12 13
14 15
16 17
23 24
25 26
27 28
Our cohort of patients showed evidence of improved mobility, as reflected
by changes in elderly mobility scale score after training in the geriatric
day hospital, with a median time to referral of 20 months from the initial
clinical presentation. Patients with PSP should be referred to a
physiotherapist and an occupational therapist for fall assessment, as well
as guidance regarding the potential need for walking aids. Relatives
should be educated to ensure close monitoring of environmental risks for
falls. The patients in our cohort showed relatively early development of
memory problems at a median duration of 9 months, which contrasted with
the mean duration of 12 months observed in another study.6 There may have
been bias in the current cohort, which recruited patients with PSP from a
geriatrics clinic whereas the patients in the previous study were
recruited from a neurology clinic.6
A previous meta-analysis showed mixed results with
regard to whether the age at onset of PSP was predictive of mortality.5 Pooled results from six studies showed no prognostic
effect of yearly increases in age at disease onset in either univariate or
multivariate analyses.5 However,
pooled results from nine studies using median age at onset as cut-off
showed a pooled HR of 1.75 (95% CI=1.32-2.32) in multivariate analysis.5 Other predictors of mortality
found in the present study, including vertical gaze palsy, dysphagia,
pneumonia, or pressure injuries, were previously reported in other
studies.6 7 8 9 10 11 12 13 14 15 16 It
remains unknown whether resolution of dysphagia in patients with PSP can
prevent pneumonia and reduce mortality. It is important for clinicians to
refer patients with PSP involving dysphagia to a speech therapist for
advice regarding food texture and appropriate swallowing posture (ie,
chin-tuck) [Table 4].29
During the clinical course of disease, approximately 40% of primary
caregivers complained of caregiver stress, which was also determined to be
a significant predictor of mortality. Patient aggression and depression
have been reported as sources of stress.4
When these positive predictors of mortality appear, clinicians should
consider discussing ACP with patients or caregivers (Table
4).
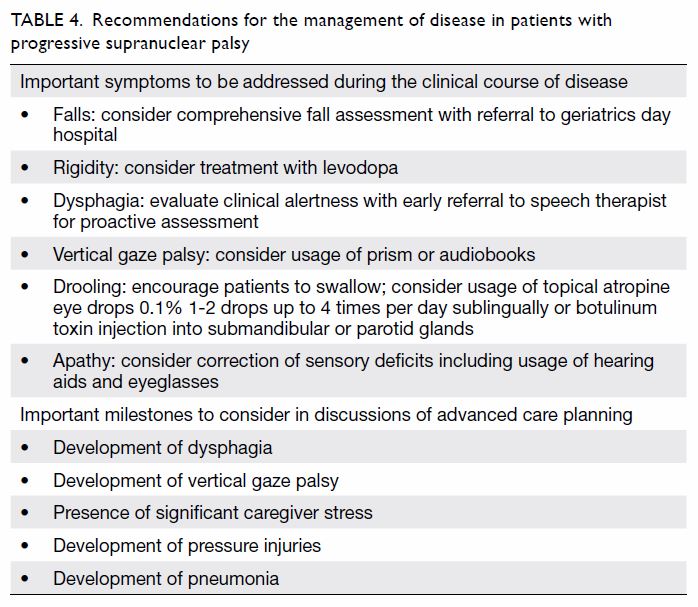
Table 4. Recommendations for the management of disease in patients with progressive supranuclear palsy
The finding of apathy as a time-dependent covariate
for prediction of mortality is notable. Recently, apathy was found to
predict survival in a cohort of 124 patients with syndromes associated
with frontotemporal lobar degeneration (including 35 patients with PSP,
mean age of 72.2 ± 8.5 years).30
The development of apathy in patients with PSP was related to brainstem,
midbrain, and frontal atrophy.30
It remains unknown whether apathy accelerates the decline to death or
indirectly signifies the degree of brainstem degeneration, including the
development of dysphagia, which is also related to greater mortality.
Future clinical trials may consider the use of therapeutic measures to
address apathy, in order to assess their impacts on the survival of
patients with PSP.
Because there is currently no disease-modifying
treatment for patients with PSP, ACP is an important component of clinical
care, for which patients and their caregivers can reach a consensus during
the clinical course of disease.4 In
addition, symptomatic care plays an important role. Symptoms relevant to
patients with PSP include dystonia, drooling, gaze palsy (also known as
reduced blinking), constipation, and apathy.4
Drooling could be managed by the administration of sublingual atropine
drops (Table 4).4
Reduced blinking could be managed by frequent application of lubricating
eyedrops. Gaze palsy could be managed by the use of prisms or audiobooks (Table 4).4
Apathy could be minimised by addressing sensory deficits, such as through
the use of eyeglasses or hearing aids (Table 4).4 Up
to 75% of patients with PSP could be discharged home after stabilisation
of symptoms.4
There are multiple limitations in the current
study. First, it was a retrospective study involving reviews of clinical
charts, and may be biased due to inconsistent documentation of symptoms.
Second, there was a limited number of patients included, none of whom had
autopsy and pathological confirmation of their diagnosis; however, we had
radiological evidence of PSP. Third, our descriptive statistical results
should be regarded as preliminary findings; only limited variables could
be included in our Cox proportional hazards modelling for survival
analysis. Notably, no specific scales were used to assess the severity of
parkinsonism or other symptoms, including response to levodopa; most of
our evaluations were subjective. Sequential C-MMSE scores were not
recorded; thus, we were unable to examine the development of dementia over
time. Fourth, we only included patients attending the geriatrics clinic;
therefore, our cohort may not be fully representative of patients with PSP
who present to most neurology clinics. Finally, the limited numbers of
patients precluded stratified analyses based on subtypes of PSP.
In conclusion, important clinical milestones,
including the development of dysphagia, vertical gaze palsy, significant
caregiver stress, pressure injuries, and pneumonia, may be used to guide
the ideal timing for discussions of ACP for patients with PSP, in order to
facilitate long-term care.
Author contributions
All authors had full access to the data,
contributed to the study, approved the final version for publication, and
take responsibility for its accuracy and integrity.
Concept or design: YF Shea, ACK Shum.
Acquisition of data: YF Shea, ACK Shum.
Analysis or interpretation of data: All authors.
Drafting of the article: YF Shea, ACK Shum.
Critical revision for important intellectual content: All authors.
Acquisition of data: YF Shea, ACK Shum.
Analysis or interpretation of data: All authors.
Drafting of the article: YF Shea, ACK Shum.
Critical revision for important intellectual content: All authors.
Conflicts of interest
The authors have no conflicts of interest to
disclose.
Funding/support
This research received no specific grant from any
funding agency in the public, commercial, or not-for-profit sectors.
Ethics approval
This retrospective study protocol was approved by
the Institutional Review Board of the University of Hong Kong/ Hospital
Authority Hong Kong West Cluster (HKU/HA HKW IRB; Ref UW 17-483) and New
Territories West Cluster Clinical and Research Ethics Committee (NTWC
CREC; Ref NTWC/CREC/17127). The requirement for informed consent was
waived by the review board.
References
1. Boxer AL, Yu JT, Golbe LI, Litvan I,
Lang AE, Höglinger GU. Advances in progressive supranuclear palsy: new
diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol
2017;16:552-63. Crossref
2. Höglinger GU, Respondek G, Stamelou M,
et al. Clinical diagnosis of progressive supranuclear palsy: The movement
disorder society criteria. Mov Disord 2017;32:853-64. Crossref
3. Luk JK, Chan FH. End-of-life care for
advanced dementia patients in residential care home—a Hong Kong
perspective. Ann Palliat Med 2018;7:359-64. Crossref
4. Wiblin L, Lee M, Burn D. Palliative care
and its emerging role in multiple system atrophy and progressive
supranuclear palsy. Parkinsonism Relat Disord 2017;34:7-14. Crossref
5. Glasmacher SA, Leigh PN, Saha RA.
Predictors of survival in progressive supranuclear palsy and multiple
system atrophy: a systematic review and meta-analysis. J Neurol Neurosurg
Psychiatry 2017;88:402-11. Crossref
6. Arena JE, Weigand SD, Whitwell JL, et
al. Progressive supranuclear palsy: progression and survival. J Neurol
2016;263:380-9. Crossref
7. Golbe LI, Davis PH, Schoenberg BS,
Duvoisin RC. Prevalence and natural history of progressive supranuclear
palsy. Neurology 1988;38:1031-4. Crossref
8. Nath U, Ben-Shlomo Y, Thomson RG, Lees
AJ, Burn DJ. Clinical features and natural history of progressive
supranuclear palsy: a clinical cohort study. Neurology 2003;60:910-6. Crossref
9. Oliveira MC, Ling H, Lees AJ, Holton JL,
De Pablo-Fernandez E, Warner TT. Association of autonomic symptoms with
disease progression and survival in progressive supranuclear palsy. J
Neurol Neurosurg Psychiatry 2019;90:555-61. Crossref
10. Cosseddu M, Benussi A, Gazzina S, et
al. Natural history and predictors of survival in progressive supranuclear
palsy. J Neurol Sci 2017;382:105-7. Crossref
11. Chiu WZ, Kaat LD, Seelaar H, et al.
Survival in progressive supranuclear palsy and frontotemporal dementia. J
Neurol Neurosurg Psychiatry 2010;81:441-5. Crossref
12. Dell’Aquila C, Zoccolella S, Cardinali
V, et al. Predictors of survival in a series of clinically diagnosed
progressive supranuclear palsy patients. Parkinsonism Relat Disord
2013;19:980-5. Crossref
13. Santacruz P, Uttl B, Litvan I, Grafman
J. Progressive supranuclear palsy: a survey of the disease course.
Neurology 1998;50:1637-47. Crossref
Litvan I, Mangone CA, McKee A, et al.
Natural history of progressive supranuclear palsy
(Steele-Richardson-Olszewski syndrome) and clinical predictors of
survival: a clinicopathological study. J Neurol Neurosurg Psychiatry
1996;60:615-20. Crossref
15. O’Sullivan SS, Massey LA, Williams DR,
et al. Clinical outcomes of progressive supranuclear palsy and multiple
system atrophy. Brain 2008;131:1362-72. Crossref
16. Papapetropoulos S, Gonzalez J, Mash
DC. Natural history of progressive supranuclear palsy: a clinicopathologic
study from a population of brain donors. Eur Neurol 2005;54:1-9. Crossref
17. Shea YF, Ha J, Chu LW. Progressive
supranuclear palsy presenting initially as semantic dementia. J Am Geriatr
Soc 2014;62:2459-60. Crossref
18. Chiu HF, Lee HC, Chung WS, Kwong PK.
Reliability and validity of Cantonese version of the Mini-Mental State
Examination: a preliminary study. J Hong Kong Col Psych 1994;4:25-8.
19. Prosser L, Canby A. Further validation
of the elderly mobility scale for measurement of mobility of hospitalized
elderly people. Clin Rehabil 1997;11:338-43. Crossref
20. Tomita S, Oeda T, Umemura A, et al.
Impact of aspiration pneumonia on the clinical course of progressive
supranuclear palsy: a retrospective cohort study. PLoS One
2015;10:e0135823. Crossref
21. Rosenbek JC, Robbins JA, Roecker EB,
Coyle JL, Wood JL. A penetration-aspiration scale. Dysphagia 1996;11:93-8.
Crossref
22. Edsberg LE, Black JM, Goldberg M,
McNichol L, Moore L, Sieggreen M. Revised national pressure ulcer advisory
panel pressure injury staging system: revised pressure injury staging
system. J Wound Ostomy Continence Nurs 2016;43:585-97. Crossref
23. Testa D, Monza D, Ferrarini M,
Soliveri P, Girotti F, Filippini G. Comparison of natural histories of
progressive supranuclear palsy and multiple system atrophy. Neurol Sci
2001;22:247-51. Crossref
24. Diroma C, Dell’Aquila C, Fraddosio A,
et al. Natural history and clinical features of progressive supranuclear
palsy: a clinical study. Neurol Sci 2003;24:176-7. Crossref
25. Goetz CG, Leurgans S, Lang AE, Litvan
I. Progression of gait, speech and swallowing deficits in progressive
supranuclear palsy. Neurology 2003;60:917-22. Crossref
26. Jecmenica-Lukic M, Petrovic IN,
Pekmezovic T, Kostic VS. Clinical outcomes of two main variants of
progressive supranuclear palsy and multiple system atrophy: a prospective
natural history study. J Neurol 2014;261:1575-83. Crossref
27. Litvan I, Kong M. Rate of decline in
progressive supranuclear palsy. Mov Disord 2014;29:463-8. Crossref
28. Ou R, Liu H, Hou Y, et al. Executive
dysfunction, behavioral changes and quality of life in Chinese patients
with progressive supranuclear palsy. J Neurol Sci 2017;380:182-6. Crossref
29. Luk JK, Chan DK. Preventing aspiration
pneumonia in older people: do we have the ‘know-how’? Hong Kong Med J
2014;20:421-7. Crossref
30. Lansdall CJ, Coyle-Gilchrist IT,
Vázquez Rodríguez P, et al. Prognostic importance of apathy in syndromes
associated with frontotemporal lobar degeneration. Neurology
2019;92:e1547-57. Crossref