Hong
Kong Med J 2017 Dec;23(6):579–85 | Epub 10 Nov 2017
DOI: 10.12809/hkmj176845
© Hong Kong Academy of Medicine. CC BY-NC-ND 4.0
ORIGINAL ARTICLE
The first case series of Chinese patients in Hong Kong
with familial Alzheimer’s disease compared with those with
biomarker-confirmed sporadic late-onset Alzheimer’s disease
YF Shea, MRCP (UK), FHKAM (Medicine)1;
LW Chu, MD, FRCP (Lond)1; SC Lee, BHS(Nursing)1;
Angel OK Chan, MD, FRCPA2
1 Division of Geriatric Medicine,
Department of Medicine, LKS Faculty of Medicine, The University of Hong
Kong, Queen Mary Hospital, Pokfulam, Hong Kong
2 Division of Clinical
Biochemistry, Department of Pathology & Clinical Biochemistry, Queen
Mary Hospital, Pokfulam, Hong Kong
Corresponding author: Dr YF Shea (elphashea@gmail.com)
 Full
paper in PDF
Full
paper in PDF
Abstract
Introduction: Patients with
familial Alzheimer’s disease are being increasingly reported in Hong
Kong. The objectives of this study were to report the clinical features
of these patients, and to compare them with those with
biomarker-confirmed sporadic late-onset Alzheimer’s disease.
Methods: All symptomatic Chinese
patients with familial Alzheimer’s disease who attended Queen Mary
Hospital, Memory Clinic between January 1998 and December 2016 were
included. Information about clinical features, baseline Mini-Mental
State Examination score, and presenting cognitive symptoms or atypical
clinical features were collected. Their clinical features were compared
with those of 12 patients with sporadic late-onset Alzheimer’s disease
with cerebrospinal fluid biomarker evidence of Alzheimer’s disease and
14 patients with late-onset Alzheimer’s disease and positive amyloid
loading on Pittsburgh compound B imaging.
Results: There were three
families with familial Alzheimer’s disease among whom eight family
members were affected. The mean (± standard deviation) age of onset and
the Mini-Mental State Examination score were 48.4 ± 7.7 years and 7.9 ±
9.2, respectively. Compared with the sporadic late-onset Alzheimer’s
disease patients, those with familial Alzheimer’s disease had an earlier
age of onset and presentation (both P<0.001) and received the correct
diagnosis later (median [interquartile range], 7.5 [5.3-14.5] vs 2
[1.0-3.3] years; P<0.001). Patients with familial disease had a lower
Mini-Mental State Examination score at presentation than those having
late-onset Alzheimer’s disease (mean, 7.9 ± 9.2 vs 17.6 ± 7.2; P=0.01).
They also had fewer delusions, and less dysphoria and irritability (0%
vs 41.7%, 0% vs 50% and 0% vs 54.2%; P=0.04, 0.01 and 0.01,
respectively). There was a trend of less frequent amnesia among patients
with familial Alzheimer’s disease compared with those having late-onset
Alzheimer’s disease (75% vs 100%; P=0.05).
Conclusion: Clinical features
differ for patients with familial Alzheimer’s disease compared with
those with late-onset Alzheimer’s disease. There is a delay in
diagnosis. Promotion of public awareness of familial Alzheimer’s disease
is much needed.
New knowledge added by this study
- There is a significant delay in the diagnosis of familial Alzheimer’s disease (FAD) in Hong Kong.
- Patients with FAD had fewer delusions and less dysphoria and irritability compared with patients with sporadic late-onset Alzheimer’s disease.
- Promotion of public awareness of FAD is much needed.
Introduction
Alzheimer’s disease (AD) is the most common cause
of dementia. It is frequently classified as early-onset AD (EOAD) if onset
is before the age of 65 years and thereafter as late-onset AD (LOAD).
Familial AD (FAD) is a special form of EOAD with an autosomal dominant
inheritance, and can be caused by mutations in presenilin (PSEN) 1
or 2 and amyloid precursor protein (APP) genes. Not all patients
with EOAD have autosomal dominant FAD, which accounts for less than 1% of
all AD.1 The first patient
diagnosed with AD by Alois Alzheimer was called Auguste Deter; she was
admitted to a psychiatric unit because of amnesia and hallucinations at
the age of 51 years.2
Deoxyribonucleic acid was extracted from a histological section of Auguste
Deter’s brain and 100 years later a heterozygous mutation p.Phe176Leu was
discovered in the PSEN1 gene.2
Unlike reports of FAD in the western population, little has been written
about this condition in the Chinese population.1
Apart from the difference in age of onset (AOO),
EOAD shows a number of differences in clinical features when compared with
LOAD. Patients with EOAD often have a non-amnestic presentation with
visuospatial dysfunction and apraxias; neuropsychologically they exhibit
dysexecutive function, and poor visuospatial and motor skills.3 Structural imaging also reveals that patients with EOAD
exhibit more frontal or temporoparietal atrophy rather than the
hippocampal atrophy seen in patients with LOAD.3
Patients with EOAD exhibit more hypometabolism in the temporoparietal
cortex while those with LOAD exhibit more hypometabolism over the medial
temporal lobe.3 Our previous
systematic review revealed that FAD patients can present with atypical
clinical features including myoclonus, seizures, cerebellar dysfunction,
spastic paraparesis, and neuropsychiatric manifestations.1 These factors may contribute to under-recognition of
EOAD or FAD among local Chinese population.
Diagnosis of FAD is clinically important for the
affected family. Genetic counselling may be offered to potential
asymptomatic carriers if desired, as they may benefit from prenatal
diagnosis and planning of personal affairs.4
Identification of asymptomatic carriers can also identify potential
candidates for future drug trials of disease-modifying agents. With
respect to preventive therapies, two clinical trials—the DIAN-TU
(Dominantly Inherited Alzheimer Network Trial Unit) and API (Alzheimer’s
Prevention Initiative)—are ongoing to test the efficacy of passive
immunotherapy among normal or mildly symptomatic FAD mutation carriers.5 6
Thus, it is important to enhance local doctors’ knowledge of FAD.
The objectives of this study were to report the
clinical features of the first case series of Chinese FAD patients in Hong
Kong, and to compare their clinical features with those of
biomarker-confirmed sporadic LOAD patients. We hypothesised that patients
with FAD had more atypical clinical features, and that this could
contribute to a delay in correct diagnosis.
Methods
Patients with familial Alzheimer’s disease
This was a retrospective case series of FAD
patients diagnosed between January 1998 and December 2016 in the memory
clinic of Queen Mary Hospital, Hong Kong. The FAD patients were identified
by reviewing the case records of all patients diagnosed with EOAD during
the study period. The study was performed in accordance with the
principles outlined in the Declaration of Helsinki. All symptomatic FAD
patients with confirmed mutations in PSEN1 or APP genes
were included. To date, no FAD family with PSEN2 has been
identified in Hong Kong. All these patients are pure Chinese. Detailed
histories were obtained from primary caregivers. All patients underwent a
physical examination, laboratory blood tests (including vitamin B12,
folic acid, and thyroid function), computed tomography (CT) or magnetic
resonance imaging (MRI) of the brain, and completed the Cantonese version
of Mini-Mental State Examination (MMSE).7
These patients fulfilled the National Institute of Neurological and
Communicative Disorders and Stroke and the Alzheimer’s Disease and Related
Disorders Association (NINCDS-ADRDA) diagnostic criteria of AD.8 In this study, AOO was defined as the age at first
appearance of symptoms that interfered with social or occupational
functioning. Age of correct diagnosis (AOCD) was defined as the age at
which diagnosis of FAD was confirmed with genetic mutation. Duration of
symptoms was defined as the difference between AOO and AOCD in years.
Initial presenting cognitive symptoms and behavioural and psychological
symptoms of dementia (BPSD) according to the Neuropsychiatric Inventory
(NPI) were specifically collected from primary caregivers and were
immediately recorded in the medical records.9
Of note, BPSD—including delusions, hallucinations, agitation, dysphoria,
anxiety, euphoria, apathy, disinhibition, irritability, and aberrant motor
behaviour—were recorded as binary variables (ie present or absent) as not
all NPI scores could be retrieved.9
Authors (SYF and LSC), who were blinded to the hypothesis, retrieved the
information related to initial presenting cognitive symptoms and BPSD.
Selection of patients
In summary, three families among whom eight
patients were affected were included in this case series. Two families
have been reported previously.10 11 For reference purposes, there
were 18 patients with EOAD and no positive family history during the study
period.
Two patients with familial Alzheimer’s disease
This family has not been reported in detail
previously. The family was referred to our memory clinic more than 10
years ago (Fig). The first case (II3; patient No. 5) was a
52-year-old woman who complained of progressive short-term memory
impairment with impaired daily function, occupational performance, and
management of personal finances. Her father (I1) and eldest brother (II1)
had been diagnosed with dementia at around 50 years of age by doctors in
China. As a result of these symptoms, her husband had divorced her, and
she received care from her friend. Single-photon emission CT of the brain
showed bilateral hypoperfusion over the frontal and temporoparietal lobes.
She consented to genetic testing and gene sequencing for known FAD
mutations, which was subsequently performed by The Tanz Centre for
Research in Neurodegenerative Diseases, University of Toronto. A
heterozygous missense mutation p.His163Arg in the PSEN1 gene was
detected. She received rivastigmine treatment. Five years later, because
of her poor drug compliance and impaired ability to carry out cooking and
housework, arrangements were made for her to live in an elderly care home.
Another patient (II4; patient No. 4) was her 46-year-old brother who was
diagnosed with dementia by another hospital. He also consented to have
genetic testing. The same heterozygous missense mutation was found.
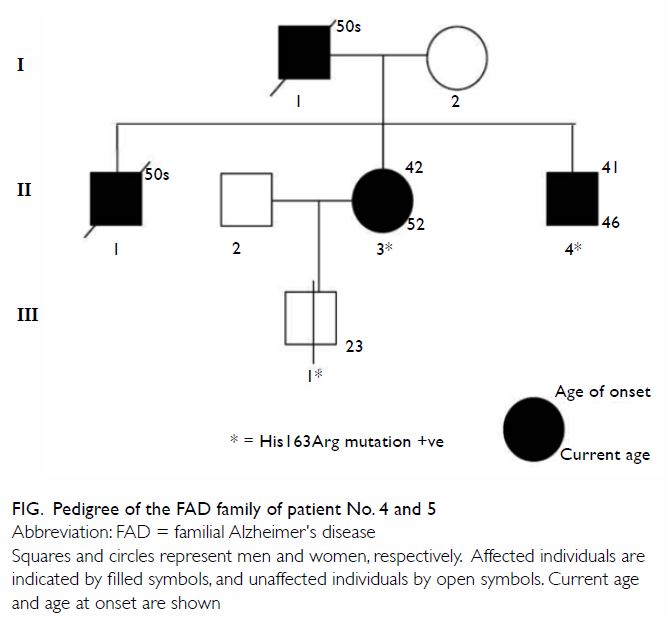
Figure. Pedigree of the FAD family of patients No.4 and 5
Late-onset Alzheimer’s disease with biomarker
confirmation
Late-onset AD is defined as AD with AOO that occurs
at or after the age of 65 years. During the study period, 12 patients with
LOAD underwent cerebrospinal fluid (CSF) examination that revealed an AD
pattern of CSF biomarkers (ie low amyloid-beta [Aβ42], and elevated total
tau and phosphorylated tau [pTau]) within 1 month of clinical assessment.12 In addition, 14 patients with
LOAD underwent 11C-Pittsburgh compound B (PIB) and 18F-2-fluoro-2-deoxy-d-glucose
(FDG) positron emission tomography (PET) within 3 months of clinical
assessment. Bilateral temporoparietal hypometabolism was evident on 18FDG
PET and positive amyloid loading on 11C-PIB (ie binding
occurred in more than one cortical brain region: frontal, parietal,
temporal, or occipital).13
Clinically, these patients also fulfilled the NINCDS-ADRDA criteria for
AD. These patients had no history of stroke and their CT or MRI brain
showed no evidence of infarcts or extensive white matter changes. For
these 26 patients with LOAD, similar clinical information including basic
demographics, AOO, AOCD (based on the availability of biomarkers’
results), disease duration, Cantonese version of MMSE, initial presenting
cortical symptoms, and BPSD was collected. For reference purposes, there
were 2480 patients with LOAD without CSF biomarkers or FDG and PIB-PET
examination during the same period of time.
Statistical analysis
Parametric variables are expressed as mean ±
standard deviation (SD). Non-parametric variables are expressed as median
with interquartile range (IQR). Chi squared test or Fisher’s exact test
were used to compare categorical variables. Independent sample t
test or Mann-Whitney U test was used to compare continuous
variables when appropriate. Statistical significance was inferred by a
two-tailed P value of <0.05. All statistical analyses were performed
using the SPSS (Windows version 18.0; SPSS Inc, Chicago [IL], US).
Results
Case series of familial Alzheimer’s disease
There were three affected families with eight
affected patients. Their clinical features are summarised in Table
1. The mean (± SD) AOO and MMSE score were 48.4 ± 7.7 years and 7.9
± 9.2, respectively. The mean duration of symptoms before genetic
diagnosis was 10.1 ± 7.1 years. Patients 1 and 3 were initially
misdiagnosed with depression and Parkinson’s disease with dementia,
respectively. The three most common presenting cognitive symptoms were
amnesia (75%), disorientation (63%), and anomia (38%).
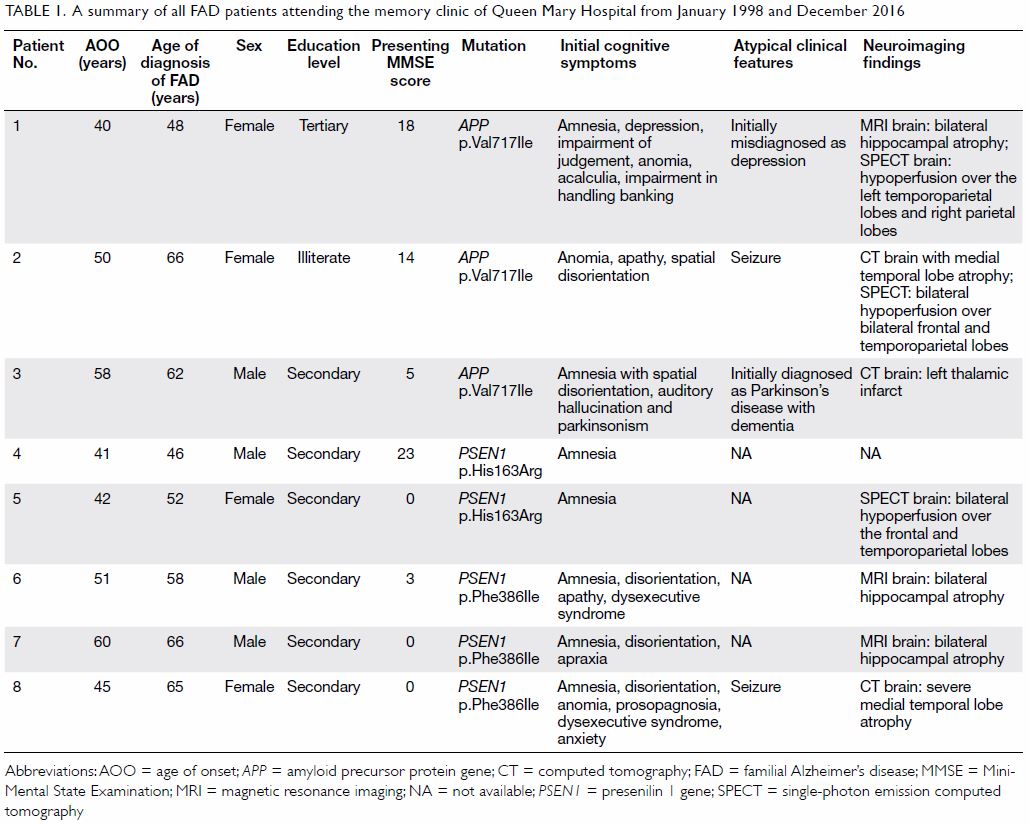
Table 1. A summary of all FAD patients attending the memory clinic of Queen Mary Hospital from January 1998 and December 2016
Comparison with late-onset Alzheimer’s disease
The comparison of demographics between FAD and LOAD
patients is summarised in Table 2. The AOO and AOCD were much earlier for FAD
than LOAD patients (48.4 ± 7.7 vs 77.9 ± 6.7 years and 57.9 ± 8.2 vs 80.7
± 6.2 years; both P<0.001). The duration of symptoms was much longer
for FAD patients than LOAD patients (median [IQR]: 7.5 [5.3-14.5] vs 2.0
[1.0-3.3] years; P<0.001). Patients with FAD had a lower presenting
MMSE score than those with LOAD (7.9 ± 9.2 vs 17.6 ± 7.2; P=0.01). More
patients with FAD had been educated to secondary level or above than LOAD
patients (P=0.001).
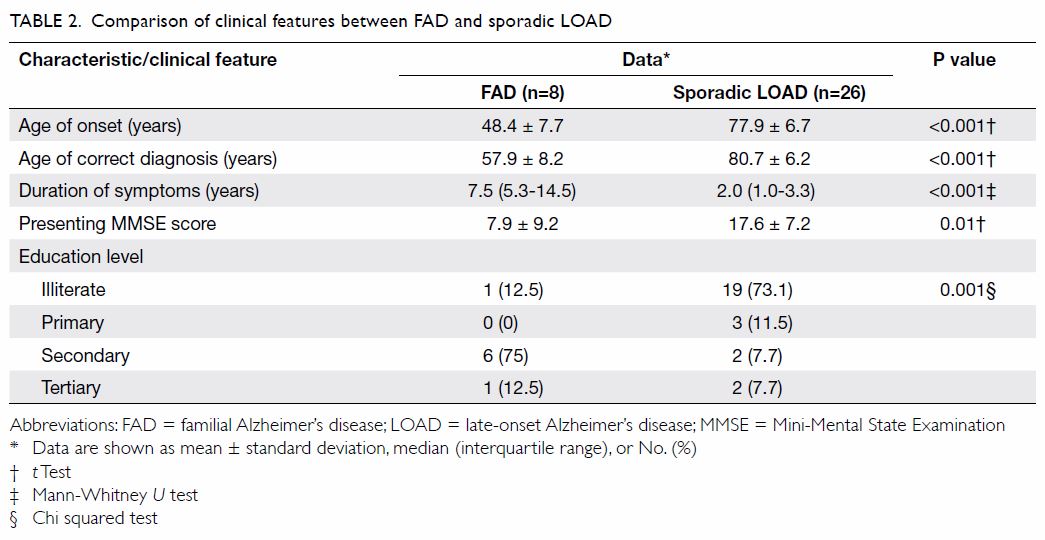
Table 2. Comparison of clinical features between FAD and sporadic LOAD
The comparison of cognitive symptoms and BPSD
between FAD and LOAD patients is summarised in Tables 3
and 4, respectively. There was a trend wherein patients
with FAD were less likely to present with amnesia (75% vs 100%; P=0.05)
than those with LOAD although it was still their main presenting cognitive
symptom. Patients with LOAD more commonly presented with delusion,
dysphoria, and irritability than FAD patients (0% vs 41.7%, 0% vs 50%, and
0% vs 54.2% respectively; P=0.04, 0.01, and 0.01, respectively).
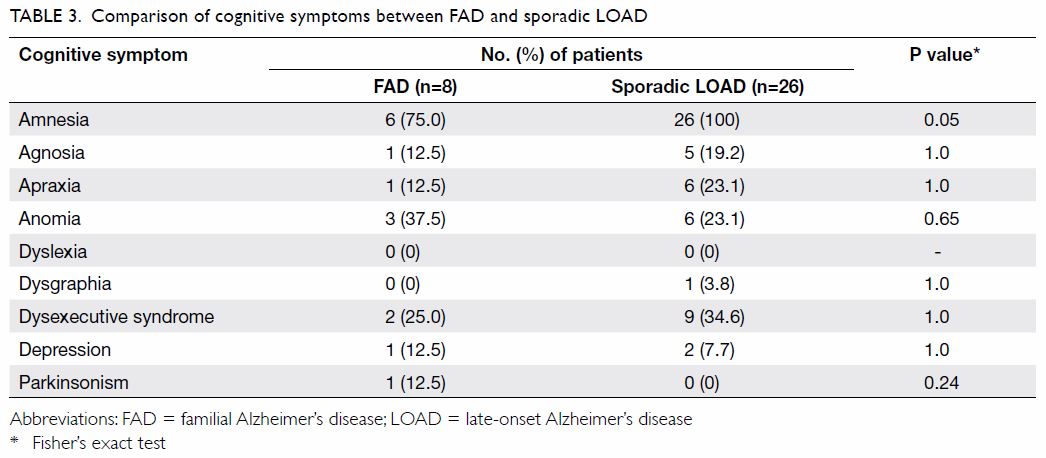
Table 3. Comparison of cognitive symptoms between FAD and sporadic LOAD
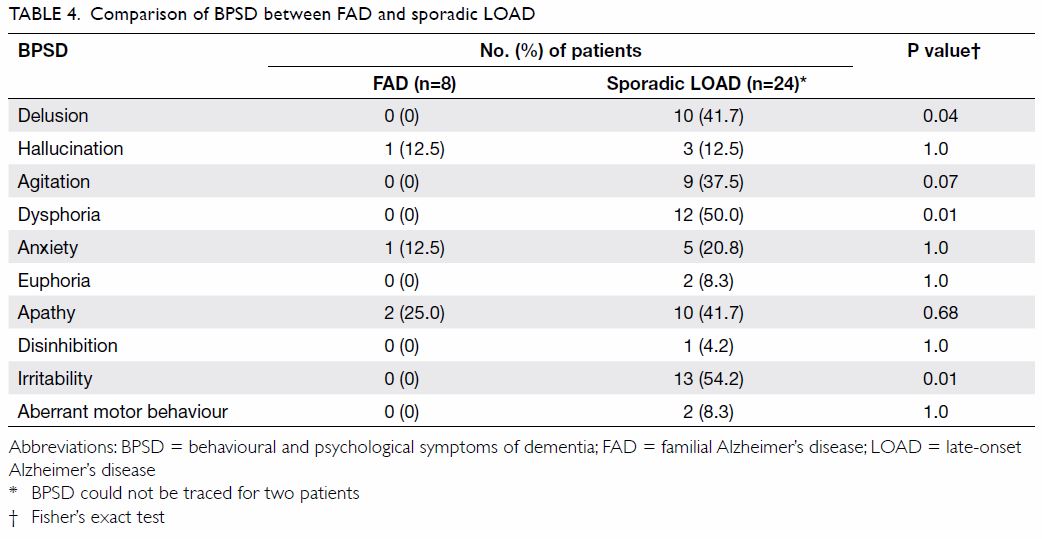
Table 4. Comparison of BPSD between FAD and sporadic LOAD
Discussion
In this case series, there was significant delay in
making a correct diagnosis of FAD among patients who presented at a late
stage of dementia compared with patients with LOAD. Patients with LOAD
more often presented with BPSD such as delusion, dysphoria, and
irritability.
There are several factors that contribute to the
delay in diagnosis and thus the late presentation of FAD patients to the
memory clinic. First, the availability of genetic tests is not well known
to local doctors. Currently doctors in public hospitals can consult with a
clinical biochemist if they encounter a family with at least two
generations having EOAD. Genetic tests can be arranged for PSEN1, APP,
and PSEN2 sequentially. Second, patients with FAD may have
atypical clinical features. In our case series, two patients were
initially misdiagnosed as depression and Parkinson’s disease with
dementia. Our previous systematic review indicated that patients with FAD
and PSEN1 mutations can present with parkinsonism, seizures,
spastic paraparesis, myoclonus, and cerebellar dysfunction.1 Chinese FAD patients with an APP mutation can
present with atypical phenotypes with a prominent psychiatric
manifestation, behavioural and language variants.1
Patients with FAD with a PSEN2 mutation can present with a later
AOO even within the same family.1
It is important for local doctors to be aware of the possibility of these
atypical clinical features in their EOAD patients, especially if there is
a positive family history of EOAD. Third, since FAD is not treatable,
genetic testing may not be considered. Nonetheless, genetic counselling is
important for patients with FAD. Asymptomatic carriers are also
potentially valuable for future clinical trials.4
5 6
In terms of cognitive symptoms, patients with FAD
tended to present slightly less frequently with amnesia than those with
LOAD, although amnesia remained their main presenting cognitive symptom.
This is in agreement with previous studies that reported EOAD patients to
have more prominent frontoparietal dysfunction than medial temporal
dysfunction.3 14 15 Our study
also identified that LOAD patients have more positive symptoms of BPSD
including delusions and irritability. Table 5 summarises the differences in BPSD between
FAD and LOAD patients in our study and in other reported studies between
EOAD and LOAD patients.16 17 18
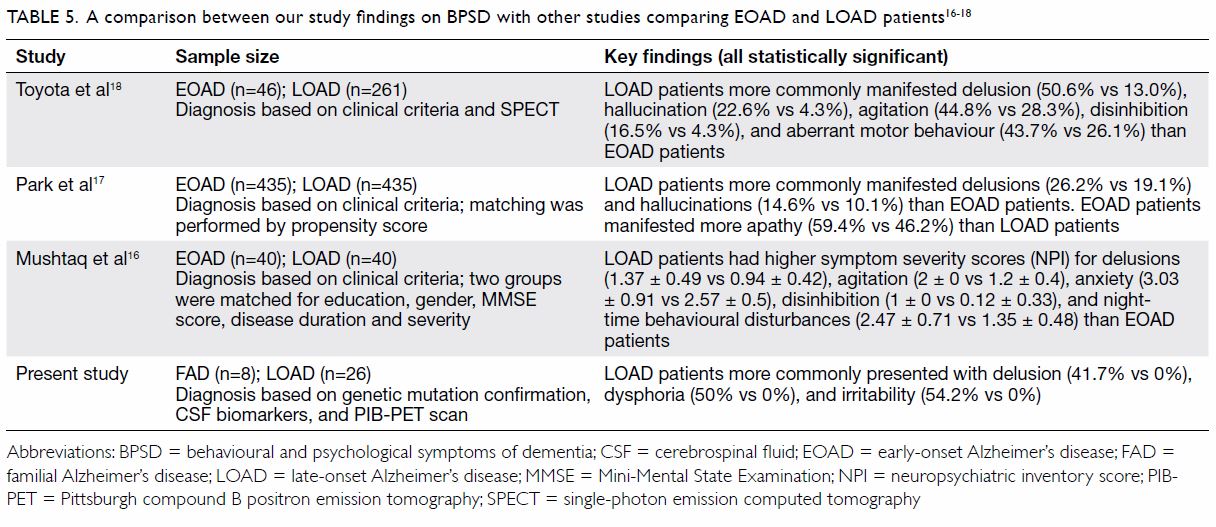
Table 5. A comparison between our study findings on BPSD with other studies comparing EOAD and LOAD patients 16 17 18
There are several potential reasons for the
different clinical features between FAD and LOAD patients. First, patients
with FAD have a genetic mutation that increases the production of Aβ42
from early on in life. This explains the much earlier AOO.19 Second, pathological studies of the brain of FAD
patients seldom noted non-AD pathological changes. On the contrary, 42% of
LOAD patients exhibited at least one other concurrent clinicopathological
diagnosis such as vascular dementia, dementia with Lewy bodies,
hippocampal sclerosis, or Pick’s disease.20
Third, amyloid plaques in LOAD patients are mostly compact or diffuse
while those in FAD patients exhibit various morphologies associated with
the specific PSEN mutation.20
Fourth, PSEN 1 and 2 form the catalytic subunit of γ-secretase and
apart from amyloid beta precursor protein, there are over 90 other
substrates upon which γ-secretase can act; this may explain the wide range
of phenotypes for FAD patients with PSEN mutations.20
The strength of the study is that the diagnoses of
FAD and LOAD were supported by genetic analyses and imaging/CSF
biomarkers, respectively. There are a number of limitations in our study.
First, FAD accounted for only a minority of EOAD cases and thus our
results may not be generalised to sporadic EOAD patients. Second, the
severity of BPSD could not be compared. In future, NPI scores should be
compared. In addition, detailed neuropsychological tests were not
performed because of the busy clinical setting in our memory clinic.
Third, the sample size is small and our results must be treated as
preliminary. Fourth, the presence or absence of symptoms depends on the
recall of primary caregivers and is subject to recall bias. Fifth,
apolipoprotein E status is an important genetic contributor to LOAD but it
was not checked in all LOAD patients in this study.4 Despite these limitations, this is the first local
study in Hong Kong to compare Chinese FAD and LOAD patients.
In summary, there are differences in clinical
features between patients with FAD, who receive a correct diagnosis much
later, and patients with LOAD. Promotion of public awareness of FAD in
Hong Kong is much needed to help those families that are affected but not
yet identified.
Declaration
All authors have disclosed no conflicts of
interest.
References
1. Shea YF, Chu LW, Chan AO, Ha J, Li Y,
Song YQ. A systematic review of familial Alzheimer’s disease: differences
in presentation of clinical features among three mutated genes and
potential ethnic differences. J Formos Med Assoc 2016;115:67-75. Crossref
2. Müller U, Winter P, Graeber MB. A
presenilin 1 mutation in the first case of Alzheimer’s disease. Lancet
Neurol 2013;12:129-30. Crossref
3. Tellechea P, Pujol N, Esteve-Belloch P,
et al. Early- and late-onset Alzheimer disease: are they the same entity
[in English, Spanish]? Neurologia 2015;pii:S0213-4853(15)00210-8.
4. Bird TD. Genetic aspects of Alzheimer
disease. Genet Med 2008;10:231-9. Crossref
5. Bateman RJ, Benzinger TL, Berry S, et
al. The DIAN-TU Next Generation Alzheimer’s prevention trial: adaptive
design and disease progression model. Alzheimers Dement 2017;13:8-19. Crossref
6. Reiman EM, Langbaum JB, Fleisher AS, et
al. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation
of presymptomatic treatments. J Alzheimers Dis 2011;26 Suppl 3:321-9.
7. Chiu FK, Lee HC, Chung WS, Kwong PK.
Reliability and validity of the Cantonese version of the Mini-Mental State
Examination—A preliminary study. J Hong Kong Coll Psychiatr
1994;4:25S-28S.
8. McKhann GM, Knopman DS, Chertkow H, et
al. The diagnosis of dementia due to Alzheimer’s disease: recommendations
from the National Institute on Aging-Alzheimer’s Association workgroups on
diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement
2011;7:263-9. Crossref
9. Cummings JL, Mega M, Gray K,
Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric
Inventory: comprehensive assessment of psychopathology in dementia.
Neurology 1994;44:2308-14. Crossref
10. Shea YF, Chan AO, Chu LW, et al. Novel
presenilin 1 mutation (p.F386I) in a Chinese family with early-onset
Alzheimer’s disease. Neurobiol Aging 2017;50:168.e9-11.
11. Shea YF, Chu LW, Chan AO, Kwan JS.
Delayed diagnosis of an old Chinese woman with familial Alzheimer’s
disease. J Formos Med Assoc 2015;114:1020-1. Crossref
12. Shea YF, Chu LW, Zhou L, et al.
Cerebrospinal fluid biomarkers of Alzheimer’s disease in Chinese patients:
a pilot study. Am J Alzheimers Dis Other Demen 2013;28:769-75. Crossref
13. Shea YF, Ha J, Lee SC, Chu LW. Impact
of (18)FDG PET and (11)C-PIB PET brain imaging on the diagnosis of
Alzheimer’s disease and other dementias in a regional memory clinic in
Hong Kong. Hong Kong Med J 2016;22:327-33. Crossref
14. Cavedo E, Pievani M, Boccardi M, et
al. Medial temporal atrophy in early and late-onset Alzheimer’s disease.
Neurobiol Aging 2014;35:2004-12. Crossref
15. Kaiser NC, Melrose RJ, Liu C, et al.
Neuropsychological and neuroimaging markers in early versus late-onset
Alzheimer’s disease. Am J Alzheimers Dis Other Demen 2012;27:520-9. Crossref
16. Mushtaq R, Pinto C, Tarfarosh SF, et
al. A comparison of the Behavioral and Psychological Symptoms of Dementia
(BPSD) in early-onset and late-onset Alzheimer’s disease—a study from
South East Asia (Kashmir, India). Cureus 2016;8:e625.
17. Park HK, Choi SH, Park SA, et al.
Cognitive profiles and neuropsychiatric symptoms in Korean early-onset
Alzheimer’s disease patients: a CREDOS study. J Alzheimers Dis
2015;44:661-73.
18. Toyota Y, Ikeda M, Shinagawa S, et al.
Comparison of behavioral and psychological symptoms in early-onset and
late-onset Alzheimer’s disease. Int J Geriatr Psychiatry 2007;22:896-901.
Crossref
19. Cacace R, Sleegers K, Van Broeckhoven
C. Molecular genetics of early-onset Alzheimer’s disease revisited.
Alzheimers Dement 2016;12:733-48. Crossref
20. Roher AE, Maarouf CL, Kokjohn TA.
Familial presenilin mutations and sporadic Alzheimer’s disease pathology:
is the assumption of biochemical equivalence justified? J Alzheimers Dis
2016;50:645-58.